Аминокислоты, пептиды и белки - Дэвени Т., Гергей Я. 1976
Газохроматографический анализ производных аминокислот. Состояние проблемы. Возможности и ограничения метода
Газовая хроматография пептидов
Метод Вейганда и др.
В данном методе в отличие от рассмотренных выше пептиды сначала защищают и затем, за некоторыми исключениями, непосредственно подвергают ГХ. Аминогруппы защищают остатком ТФА, а карбоксильную группу блокируют в процессе этерификации метанолом. На летучесть ТФА-соединений уже неоднократно указывалось; с помощью возгонки в высоком вакууме было достигнуто разделение соответствующих производных аминокислот, ди-, три- и более крупных пептидов [107].
Для ГХ этих соединений нужно было прежде всего решить проблемы, связанные с термостабильностью колонок и подобрать метод разделения. Из-за более высокой, чем у аминокислот, полярности для пептидов требуется, как правило, температура более 200° С. Не касаясь вопроса о стабильности фаз, отметим, что применение в данном случае полярных жидких фаз, например реоплекса или полифенилового эфира, довольно ограничено. Для сравнительно простых дипептидов их использование дало хорошие результаты, но если учесть, что при этом число разделяемых соединений было мало, вряд ли можно надеяться на более широкое применение этой методики в аналитических работах по установлению последовательности аминокислот. Удовлетворительными разделяющими фазами оказались неполярные силиконовые каучуки SE 30 и SE 52; менее выгоден апиезон L.
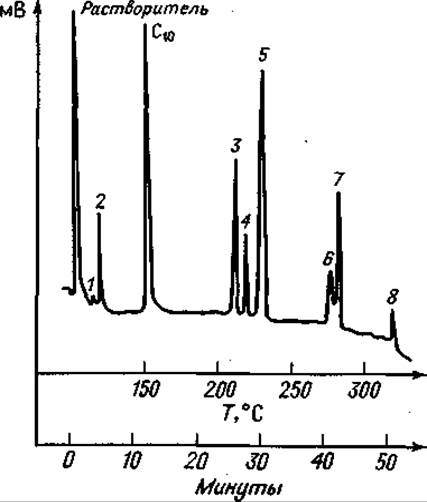
Фиг. 75. Газовая хроматограмма частичного гидролизата Лей-Лей-Вал-Вал после этерификации и трифторацетилирования.
Колонка длиной 1,8 м с 20% SE 30 на диатопорте W скорость потока Не 62 мл/мин. Сначала режим изометрический (150°С); после выхода С10 — метилового эфира каприновой кислоты — линейное программирование температуры с градиентом 5 град/мин. 1 — Вал; 2 — Лей; 3 — Вал-Вал; 4 — Лей-Вал; 5 — Лей-Лей; 6 — Лей-Вал-Вал; 7 — Лей-Лей-Вал; 8 — Лей-Лей-Вал-Вал.
При использовании подобного метода разделения для анализа последовательности аминокислот избирательность разделяющих фаз не играет большой роли, если анализируют не очень большое число соединений; решающим в данном случае будет число компонентов, детектируемых на одной колонке. Так как тетрапептиды можно обнаружить в газовом хроматографе только в исключительных случаях, наибольшее внимание неизбежно сосредоточивается на наиболее полной идентификации ди- и трипептидов. В соответствующих работах показано, что ее можно осуществить только с вышеупомянутыми фазами.
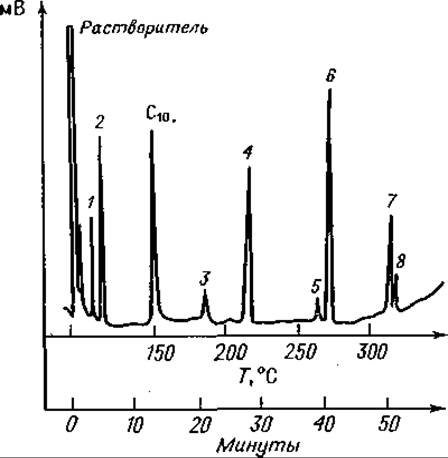
Фиг. 76. Газовая хроматограмма частичного кислотного гидролизата Лей- Фен-Вал-Вал после этерификации и трифторацетилирования.
Колонка длиной 1,8 м с 20% SE 30 на диатопорте W; скорость потока Не 62 мл/мин. Сначала режим изотермический (150°С); после выхода С10 — метилового эфира каприновой кислоты — линейное программирование температуры с градиентом 5 град/мин. 1 — Вал; 2 — Лей; 3 — Фен; 4 — Вал-Вал; 5 — Фен-Вал; 6 — Лей-Фен; 7 — Фен-Вал-Вал; 8 — Лей-Фен-Вал.
Как уже отмечалось, поскольку в смеси находится большое число различных соединений, не приходится рассчитывать на полное разделение компонентов, тем более, что разделяющие фазы обладают относительно слабой избирательностью. В связи с этим целесообразно анализировать не слишком длинные пептиды после проведения их частичного гидролиза. Например, при расщеплении декапептида в наиболее благоприятных условиях получается 9 дипептидов, 8 трипептидов и т. д. Если данный пептид состоит из 10 различных аминокислот, которые все детектируются при ГХ, тогда необходимо разделить смесь, состоящую из 27 соединений, не считая тетрапептидов, которые, вероятно, тоже могут быть обнаружены. Все возможные короткие фрагменты, разумеется, идентифицировать довольно трудно. Указанная область размеров длины цепи (от аминокислоты до тетрапептида) является, несомненно, предельной для возможностей этого метода.
Неудовлетворительное разделение, однако, пытаются компенсировать с помощью масс-спектрометрии. Было показано, что на масс-спектрометре полностью идентифицируются смеси простых пептидов [123], причем наложение пиков ни в коей мере не исключает идентификации пептидных производных.
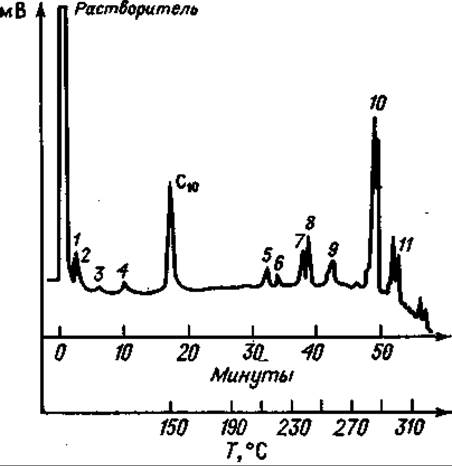
Фиг. 77. Газовая хроматограмма частичного кислотного гидролизата окситоцина после десульфирования и триметилсилилирования.

Колонка длиной 13 м с 10% SE 30 на диатопорте Р; скорость потока Не 62 мл/мии. Сначала режим изотермический (150°С); после выхода С10 — метилового эфира каприновой кислоты (стандарт) — линейное программирование температуры с градиентом 5 град/мнн. 1 — Ала; 2 — Гли; 3 — Лей; 4 — Асп; 5 — Лей-Гли; 6 — Ала-Про; 7 — Тнр; 8 — Про-Лей; 9 — Иле-Глу; (?) Глу-Асп; 10 — Ала-Тир; 11 — Тнр-Иле.
Вполне понятно, что у пептидов различия в полярности и летучести больше, чем у аминокислот. Пептиды элюируются в гораздо более широком интервале удерживаемых объемов и изотермически можно разделить лишь очень небольшую часть гидролизата. При выборе подходящих колонок и условий реакции можно было бы получить хорошее разделение и в этой части гидролизата — естественно, с относительно большими затратами (и потерями) вещества. При такой методике большая часть взятого на анализ гидролизата неизбежно теряется из-за плохого разделения (с более летучими компонентами) или неудовлетворительного элюирования (с менее летучими веществами). А так как закономерности выделения и очистки больших пептидов еще недостаточно изучены, следует избегать любых потерь. Разделение частичного гидролизата лучше проводить в одном аналитическом опыте, тогда при нанесении одинаковых количеств вещества гораздо проще проводить повторный анализ всей смеси в процессе многостадийного изотермического исследования, а компоненты смеси при этом можно надежно идентифицировать с помощью подходящего контроля.
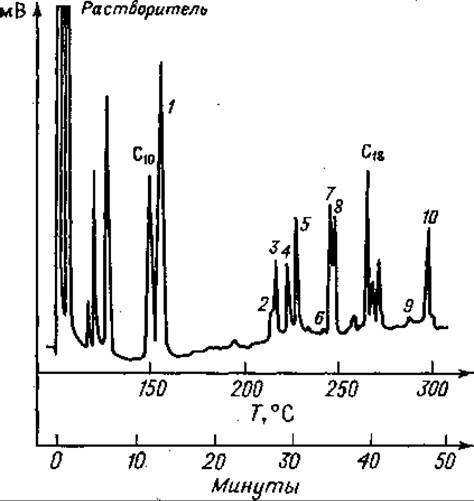
Фиг. 78. Газовая хроматограмма частичного кислотного гидролизата А-цепи инсулина после этерификации и трифторацетилироваиия.
Колонка длиной 1,8 м с 10% SE 30 иа диатопорте Р; скорость потока Не 66 мл/мин. Сначала режим изотермический (150°С); после выхода С10 — метилового эфира каприновой кислоты — линейное программирование температуры с градиентом 5 град/ми и, 1 — Глу; 2 — Сер-Вал; 3 — Иле-Вал; 4 — Сер-Лей; 5 — Тир; 6 — Вал-Глу; 7 — Глу-Лей; 8 — Лей-Глу; 9 — Лей-Тир; 10 — Глу-Глу; С18 — метиловый эфир стеариновой кислоты (стандарт).
При использовании режима с программированием температуры подобные смеси легко разделяются в одном аналитическом опыте, как видно из хроматограмм, представленных на фиг. 75—80. Отдельные группы соединений — аминокислоты, ди-, три- и тетрапептиды — выходят в различных температурных интервалах, что позволяет, несмотря на некоторое перекрывание (например, аминокислоты с большим молекулярным весом начинают выходить вместе с дипептидами), обнаруживать по виду хроматограмм качественные различия, характерные для разных последовательностей, подобно методу “отпечатков пальцев” [39]. Чтобы применять ГХ для определения последовательности, необходимо знать, какие комбинации аминокислот в ди-, три- и более длинных пептидах можно детектировать. В связи с тем что число таких сочетаний очень велико, а задача синтеза пептидов, особенно длинных, достаточно сложна, не приходится рассчитывать на получение исчерпывающих данных. Для идентификации три- и тетрапептидов выбор стандартных образцов особенно труден. Однако поведение самих аминокислот в процессе ГХ может дать существенную информацию, что позволяет избежать бесполезных экспериментов.
Превращение пептидов в N-ТФА-метиловые эфиры происходит совершенно аналогично аминокислотам: сначала их переводят в хлоргидраты метиловых эфиров, а затем трифторацетилируют. Если индивидуальные вещества не собираются определять количественно (в связи с тем, что расщепление различных пептидных связей зависит и от метода деградации, и от самой последовательности), то желательно исчерпывающее превращение, так как при этом образуется меньше продуктов расщепления. В ходе этерификации следует соблюдать предосторожности, особенно при работе с ферментативным гидролизатом, чтобы предотвратить дополнительное расщепление лабильных связей под действием кислоты. 24-часовая инкубация в 0,1 н. метанольном растворе НСl при комнатной температуре удовлетворяет необходимым условиям [15].
Трифторацетилирование избытком метилового эфира трифторуксусной кислоты в абсолютном метаноле при комнатной температуре с добавлением триэтиламина приводило к различным выходам при превращениях аминокислот [109]. Риск образования дикетопиперазинов; характерного для эфиров дипептидов, можно уменьшить при добавлении большого избытка ацилирующего агента [119]. Дикетопиперазины тоже можно подвергать газовой хроматографии, но они дают сильно асимметричные пики и поэтому непригодны для анализа последовательности [121]. Трифторацетилирование хлор гидратов пептидов можно проводить также в реакции с трифторуксусным ангидридом [119]. Хотя при этом существует опасность дополнительного расщепления пептидных связей, соответствующие продукты образуются с таким низким выходом, что это не может повлиять на последующий анализ [111]. При комнатной температуре ацилирование ангидридом протекает быстрее и более полно. Как и в методе Бимана, эфиры N-ТФА-пептидов хорошо отделяются от непрореагировавшей части молекул экстракцией кислотой и водой. При такой процедуре гидролизуются О-ТФА-производные, образовавшиеся в ходе реакции с ангидридом. Полученную смесь защищенных пептидов вносят в подходящий растворитель, например тетрагидрофуран, этилацетат или метанол, и подвергают фракционированию.
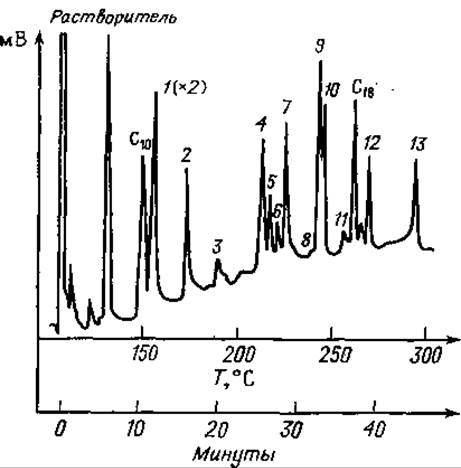
Фиг. 79. Газовая хроматограмма частичного кислотного гидролизата А-цепи инсулина после этерификации, трифторацетилирования и десульфирования.
Разделение проводили на приборе, схема которого представлена на фиг. 72.
С10 — метиловый эфир каприиовой кислоты; 1 — Глу; 2 — Ала-Ала; 3 — Вал-Ала; 4 — Сер-Вал+Иле-Вал; 5 — Ала-Асп; 6—Сер-Лей; 7 — Тир+Глу-Ала; 8 — Вал-Глу; 9 — Глу- Лей; 10 — Лей-Глу; 11 — Глу-Асп; 12 — Тир-Ала; 12 — Глу-Глу; С18 — метиловый эфир стеариновой кислоты.
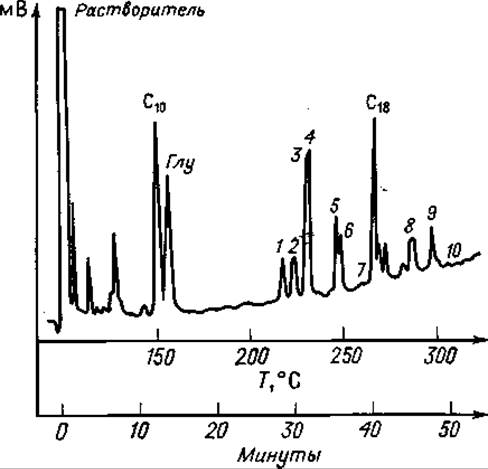
Фиг. 80. Газовая хроматограмма частичного кислотного гидролизата А-цепи инсулина после этерификации, трифторацетилирования и триметилсилилирования.
Разделение проводили на приборе, схема которого представлена иа фиг. 72.
С10 — метиловый эфир каприновой кислоты; 1 — Иле-Вал; 2 — Сер-Вал; 3 —Тир; 4— Сер- Лей, (?) Вал-Глу; 5 — Глу-Лей; 6—Лей-Глу; 7 — Глу-Асп (?); 8 — Лей-Тир; 9 — Глу-Глу; 10 — Тир-Глу; С18 — метиловый эфир стеариновой кислоты.
ГХ ди-, три-, и, возможно, многих тетрапептидов, состоящих из аминокислот Ала, Вал, Лей, Иле и Про, не вызывает никаких затруднений. Поскольку у Гли нет боковой цепи, пики соответствующих дипептидов часто имеют “хвосты”. Это явление, вероятно, вызвано эффектами адсорбции; ими же, по-видимому, объясняется неполное элюирование N-ТФА-Гли-Гли-Гли-ОСН3 [19]. С Мет были исследованы только дипептиды, а с Фен — также и трипептиды. Хотя, как следует из поведения N-ТФА-Ала-Фен-Фен-ОСН3, трипептиды, состоящие из аминокислот с высоким молекулярным весом, элюируются не всегда, при использовании коротких колонок с малым количеством жидкой фазы можно получить удовлетворительные результаты.
Что касается полифункциональных аминокислот, то дипептиды, содержащие Глу, Лиз и Орн, не представляют трудностей ни для получения производных, ни для хроматографии. Дополнительные функциональные группы защищают в ходе обычного получения производных. Однако у дипептидов, содержащих Лиз и Орн, в результате адсорбции наблюдается сильное размывание пиков на колонках, содержащих менее 5% жидкой фазы [122]. Изомерные а- и ß-пептиды, содержащие Глу, при ГХ разделяются [114, 121]. В аналогичных исследованиях метиловых эфиров N-ТФА-производных пептидов, содержащих Асп, при нагревании обнаружено образование циклических имидов, зависящее от температуры системы и более заметное для ß-пептида [106]. Отделить имиды от соответствующих а-пептидов можно только на капиллярных колонках. Возможно, при прямом внесении образца в колонку подобных реакций удалось бы избежать. Трипептиды, в которые входят указанные выше аминокислоты, до сих пор не исследованы.
Сер и Тре из-за термического ß-элиминирования во время внесения образца при высокой температуре могут давать несколько сигналов. Наилучшим методом защиты этих аминокислот оказалось триметилсилилирование гексаметилдисилазаном [121]. Тирозинсодержащие дипептиды в результате О-триметилсилилирования дают острые симметричные пики. Эти соединения могут быть приготовлены при нагревании в течение 30 мин в избытке гексаметилдисилазана после этерификации и трифторацетилирования.
До сих пор не удалось обнаружить дипептиды, содержащие Цис и Цис2. Цис особенно склонен к ß-элиминированию, на что уже указывалось в разделе об аминокислотах. Из-за отсутствия подходящих S-защитных групп были вынуждены прибегнуть к десульфированию на никеле Ренея [121]. Согласно этому методу, эфиры защищенных пептидов десульфируются на никеле Ренея в процессе продолжительного нагревания в 90%-ном водном метаноле. В результате из цистеиновых и цистиновых соединений образуются соответствующие аланиновые пептиды, которые можно отличить от аланиновых пептидов, имеющихся в исходной молекуле, с помощью D2О. Аналогично методике Бимана, SH-группа замещается на дейтерий, что обеспечивает идентификацию компонентов в масс-спектрометре. Одновременно с Цис десульфируются метиониновые соединения — они превращаются в пептиды а-аминомасляной кислоты, которые легко детектируются в газовом хроматографе.
Соединения с Apr, Гис и Три до сих пор исследованы очень мало. Сложные дипептиды, содержащие эти аминокислоты, вряд ли можно детектировать. Для Apr существует возможность его превращения в Орн с помощью аргиназы [30]. Это может оказаться полезным, так как после триптического гидролиза больших пептидов благодаря его специфичности часть остатков Apr будет находиться в С-концевом положении фрагментов. При обработке этих пептвдов аргиназой соответствующие С-концевые аминокислотные остатки превращаются в Орн, что делает пептиды доступными для ГХ. После частичного кислотного гидролиза пептиды, содержащие Три, практически не образуются, поскольку во время этой процедуры происходит разрушение Три.
Последовательность стадий вышеописанного анализа можно схематически суммировать следующим образом: 1) частичный гидролиз, 2) этерификация, 3) трифторацетилирование ГХ-анализ, 4) десульфирование ГХ-анализ, 5) триметилсилилирование ГХ-анализ.
Из сравнения результатов трех ГХ-анализов можно очень точно определить положение пептидов, содержащих оксиаминокислоты, Цис и Цис2. После десульфирования одни пики исчезают, а другие появляются — они. принадлежат серусодержащим пептидам. Если в исходном образце Мет отсутствует, то это должны быть цистеиновые (цистиновые) пептиды, которые таким образом легко идентифицируются. Та же ситуация наблюдается при количественном триметилсилилировании; при этом выявляются соединения, содержащие Сер, Тре, Опр и Тир. Производные дипептидов, содержащих Опр и Тир, элюируются при относительно высоких температурах. Таким образом, к определенным выводам можно прийти даже на основании интервала удерживаемых объемов.
В заключение можно сказать, что с помощью ГХ можно идентифицировать в виде дипептидов большинство природных аминокислот. Возможность детектирования многих аминокислот даже в виде трипептидов, а в исключительных случаях и тетрапептидов, дает в руки исследователя быстрый и относительно несложный метод определения последовательности олигопептидов средней длины. ГХ применяется сейчас в основном как быстрый и эффективный метод разделения. Более полное использование достоинств ГХ достигается при комбинировании с масс-спектрометрией [68, 106]; оно уже было с успехом применено для анализа смесей жирных кислот [75]. Весьма обещающим является использование специально сконструированных ионных источников, в результате чего становится излишним присоединение к газовому хроматографу детектора [13].
Фиг. 75—80 иллюстрируют характер ГХ N-ТФА-метиловых эфиров пептидов различной длины, которые были подвергнуты частичному кислотному гидролизу или метанолизу.
Как показано на фиг. 75 и 76, если анализируют тетрапептид, можно обнаружить все возможные фрагменты. В одном случае удается даже идентифицировать исходный пептид. На фиг. 77 представлена хроматограмма частичного гидролизата окситоцина, который в соответствии с вышеприведенной схемой десульфировали и триметилсилилировали. Были идентифицированы все возможные дипептиды, в том числе и соединения, содержащие Асп, которые образовывались при расщеплении лишь в небольшом количестве. Сравнивая различные газовые хроматограммы, можно сделать заключение о присутствии последовательностей, содержащих Цис (или Цис2) и оксиаминокислоты. На фиг. 78, 79 и 80 представлены газовые хроматограммы частичного гидролизата A-цепи инсулина, полученные на разных стадиях рассмотренных ранее превращений. После десульфирования, как видно из сравнения фиг. 78 и 79, появляются новые сигналы 2, 3 и 5, тогда как пики 7 и 12 становятся более широкими. Во всех этих случаях образуются соединения с Ала. Аналогичным образом после триметилсилилирования находят на фиг. 80 новые пики 2, 3, 4,8 и 10, а сигналы 2,4, 5 а 8, имеющиеся на фиг. 78, — исчезают. Эти пики соответствуют пептидам А-цепи, содержащим Сер и Тре, причем последние элюируются при относительно высоких температурах удерживания. После силилирования пики выходят при более высоких температурах элюирования. Как и в случае других пептидов, после кислотной деградации можно было обнаружить все ожидаемые дипептиды, за исключением дипептидов, содержащих Асп. Неидентифицированные пики, возможно, принадлежат трипептидам.
На всех рассмотренных хроматограммах компоненты идентифицировали с помощью стандартов, поскольку не было возможности для проведения масс-спектрометрического анализа.
Получаемая методом ГХ информация об исходной пептидной цепи, особенно для такой относительно большой молекулы, как A-цепь инсулина, не достаточна для определения полной последовательности. Для этого дополнительно должны быть привлечены различные методики расщепления. Тем не менее совершенно очевидно, что ГХ, главным образом как метод разделения, благодаря быстроте, малым количествам вещества, необходимого для анализа, высокой разрешающей способности обладает большими достоинствами при исследованиях в этой области. Нельзя закончить эту главу о ГХ, не упомянув о дальнейших возможностях ее применения. Так, в пептидной химии с ее помощью можно просто и быстро получить ответ на ряд вопросов. В качестве примера назовем раскрытие внутренних ангидридов N-ТФА-Глу и N-ТФА-Асп аминоэфирами, при котором образуются изомерные пептиды в различных соотношениях [106, 114].
Большое значение приобрело разделение диастереоизомерных метиловых эфиров N-ТФА-дипептидов [115]. Его проводили количественно в капиллярных колонках для большого числа пептидов различной последовательности [124]. Это разделение является первым точным и надежным методом определения рацемизации в пептидном синтезе. Наряду с разнообразными N-ациламинокислотами при изучении оптимальных условий различных реакций пептидного синтеза могут найти применение N-ацилпептиды, если синтезированные пептиды перед разделением диастереоизомеров подвергать частичному гидролизу [69, 124]. На фиг. 81 показана хроматограмма частичного кислотного гидролизата не полностью рацемизованного синтетического тетрапептида. В данных условиях, помимо аминокислот, были обнаружены только дипептиды, входящие в исходную последовательность. На фиг. 77 двойной пик тирозинового дипептида также свидетельствует о частичной рацемизации Тир. Из этого последнего примера видно, что в определенных случаях можно быстро и без затруднений контролировать и определять степень оптической чистоты пептидов.
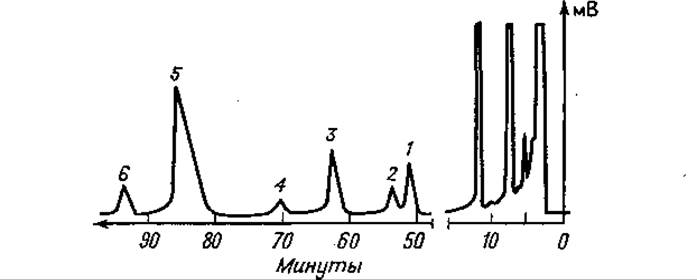
Фиг. 81. Газовая хроматограмма частичного кислотного гидролизата не полностью рацемизованного тетрапептида Лей-Фен-Ала-Фен после этерификации и трифторацетилирования.
Стальной капилляр длиной 50 м содержит полифениловый эфир OS 138; 220°С; скорость потока N2— 2,1 мл/мин. 1 — L-Фен-L-Ала; 2 — L-Фен-О-Ала; 3—L-Ала-L-Феи; 4 — D-Ала-L-Феи; 5 — L-Лeй-L-Феи; б — L-Лей-D-Фен.
Цитированная литература
1. Ackman R. G., J. of Gas Chromatogr., 2, 173—179 (1964).
2. Anders К., Gas-Chromatographie, pp. 1—22 (ed. Struppe H. G., Angele H. P., Akademie Verlag, Berlin 1964), 1963.
3. Baraud J., Bull. Soc. Chim. France, 754 (1960).
4. Bayer E., Desty D. H., Gas Chromatography, p. 333, Butterworth, London, 1958.
5. Bayer E., Born F., Reuther K. H., Angew, Chem., 69, 640 (1957).
6. Biemann K., Gapp F., Seibl J., J. Am. Chem. Soc., 81, 2274 (1959).
7. Biemann K., Vetter W., Biochem. Biophys. Res. Commun., 3, 578 (1960).
8. Bier М., Teitelbaum C., Ann. N. Y. Acad. Sci., 72, 641 (1959).
9. Birkofer L., Ritter A., Chem. Ber., 93, 424 (1950).
10. Blau К., Darbre A., Biochem. J., 88, 8P (1963).
11. Blau K., Darbre A., J. Chromatogr., 17, 445 (1965).
12. Boissonnas R. A., Gutimann S., Huguenin R. L., Jaquenoud P. A., Sandrin E., Helv. Chim. Acta, 41, 1867 (1958).
13. Вrunnёе C., Jenckel L., Kronenberger К., Z. Anal. Chem., 197, 42 (1963).
14. Calvin М., Schallenberg E., J. Am. Chem. Soc., 77, 2779 (1955).
15. Chibnall А. C., Mangan J. L., Rees M. W., Biochem. J., 68, 114 (1958).
16. Gruickshank P. A., Sheehan J. C., Anal. Chem., 36, 1191 (1964).
17. Darbre А., Личное сообщение.
18. Darbre A., Blau К., Biochem. J., 88, 8P (1963).
19. Darbre A., Blau K., J. Chromatogr., 17, 31 (1965).
20. Dorsey J. A., Hunt R. H., O’Neal M. J., Anal. Chem., 35, 511—515 (1963).
21. Eck R. V., Nature, 193, 241 (1962).
22. Edman P., Acta Chem. Scand., 4, 283 (1950).
23. Engelhardt K., Dissertation, Techn. Hochschule (Technical University), München, 1963.
24. Ettre L. S., Open Tubular Columns in Gas Chromatography, Plenum Press, New York, 1965.
25. Evans R. М., Mfg. Chemists Assoc., 32, 512 (1961).
26. Fischer E., Ber. Deut. Chem. Ges., 34, 433 (1901).
27. Gehrke C. W., Lamkin W. М., Chem. Eng. News, 42, Nr. 37, 62 (1964).
28. Giaccobbо Н., Simon W., Pharm. Acta Helv., 39, 162 (1964).
29. Graf J., Wein J. P., Winitz M., Fed. Proc., 22, Part I, 244 (1963).
30. Grassmann W., Hermann H., Janowsky O., Z. Physiol. Chem., 312, 273 (1958).
31. Greenstein J. P., Winitz A4., Chemistry of the Amino Acids, John Wiley and Sons, New York, p. 925, 1961.
32. Hagen P., Black W., Fed. Proc., 23, 371 (1964).
33. Henneberg D., Z. Anal. Chem., 183, 12—23 (1961).
34. Heyns K., Grützmacher H. F., Tetrahedron 1761; Liebings Ann. Chem., 669, 189 (1963).
35. Hoffter D., Dissertation Techn. Hochschule (Technical University), München, 1962.
36. Horning E. C., Maddock К. C., Anthony К. V., van den Heuvel W. J. A., Anal. Chem., 35, 526 (1963).
37. Hunter I. R., Dimick К. P., Cobse J. W., Chem. Inds, 294 (1956).
38. Ikekawa N., J. Biochem. (Japan), 54, 279(1963).
39. Ingram V. M., Nature, 178, 792 (1956).
40. Ishii S., Witkop B., J. Am. Chem. Soc., 85, 1832 (1963).
41. Jonak J., Nature, 185, 685 (1960).
42. Johnson D. E., Scott S. J., Meister A., Anal. Chem., 33, 669 (1961).
43. Kaelicke J., Dissertation. Techn. Universität (Technical University), Berlin, 1958.
44. Raiser R., Chromatographie in der Gasphase. Bd. II. Kapillar-Chromatographie. Bibliographisches Institut, Mannheim, 2nd ed., 1966.
45. Kaiser R., Chromatographie in der Gasphase. Bd. III. Tables pp. 78—116, Bibliographisches Institut, Mannheim, 2nd ed., 1968.
46. Kaiser R., Z. Anal. Chem., 205, 284—298 (1964).
47. Kaiser R, Chromatographie in der Gasphase. Bd. I. Gas-Chromatographie, pp. 58—59, Bibliographisches Institut, Mannheim, 1965.
48. Kaiser R., Chromatographie in der Gasphase. Bd. IV. Quantitative Auswertung. Bibliographisches Institut, Mannheim, 1965.
49. Kaiser R., Chromatographie in der Gasphase. Bd. IV, pp. 26—52, Bibliographisches Institut, Mannheim, 1965.
50. Karrer P., Nicolaus B. J. R., Helv. Chim. Acta, 35, 1581 (1952).
51. Karrer P., Portmann P., Suber A4., Helv. Chim. Acta, 32, 1156 (1949).
52. Koroats E., Wehrli A., Helv. Chim. Acta, 42, 2709 (1959).
53. Lamkin W. W., Gehrke C. IF., Anal. Chem., 37, 385 (1965).
54. Landowne R. A., Lipsky S. R., Nature, 199, 141 (1963).
55. Langheld K., Ber. Deut. Chem. Ges., 42, 2360 (1909).
56. Liberii A., Desty D. H., Gas Chromatography, Butterworth, London, p. 341, 1958.
57. Lorette N. B., Brown J. H., Jr., J. Org. Chem., 24, 261 (1959).
58. Losse G., Losse A., Stock J., Z. Naturforsch., 17b, 785 (1962).
59. Makisumi S., Nicholls C. H., Saroff H. A., J. Chromatogr., 11, 327 (1963).
60. Makisumi S., Saroff H. A., J. Gas Chromatogr., 3 , 21 (1965).
61. McCaldin D. J., Chem. Revs., 60, 39 (1960).
62. Melamed N., Renard R., J. Chromatogr., 4, 339 (1960).
63. Meyer D. M., Jutisz M., Bull. Soc. Chim. France, 1211 (1957).
64. Morita K., Irreverse F., Witkop B., J. Am. Chem. Soc., 85, 2832 (1963).
65. Nicholls С. H., Makisumi S., Saroff H. A., J. Chromatogr., 11, 327 (1963).
66. Pisano J. J., van der Heuvel E. J. A., Horning E. C., Biochem. Biophys. Res. Commun., 7, 82 (1962).
67. Prox A., Dissertation. Tecnn. Universität, Berlin, 1959.
68. Prox A., Weygand F., in: Peptides, Proc. of the 8th Europ. Peptide-Sympos. Nordwijk, the Netherlands, Sept. 1966, ed. by Beyerman H. C., van de Linde A., van den Brink M., North Holland, p. 158, 1967.
69. Prox A., Weygand F., König W., Schmidhammer L., Proceedings of the 6th Europ. Peptide-Symposium, Athens, 1963, Pergamon Press, Oxford, p. 139, 1966.
70. Rühlmann K., Giesecke W., Angew. chem., 73, 113 (1961).
71. Rühlmann К., Michael G., Chem. Ber., 94, 1876 (1961).
72. Rühlmann К., Michael G., Gas-Chromatographie, pp. 221—229, 1963.
73. Rühlmann K., Michael G., J. Gas Chromatogr., I, 18 (1963).
74. Ruttenberg M. A., King T. P., Craig L. C., Biochem. J., 3, 758 (1964).
75. Ryhage R., Anal. Chem., 36, 759 (1964).
76. Sanger F., Biochem. J., 39, 507 (1945).
77. Saroff H. A., Karmen A., Anal. Biochem., 1, 344 (1960).
78. Saroff H. A., Karmen A., Healy J. A., J. Chromatogr., 9, 122 (1962).
79. Shaw P. D., Anal. Chem., 35, 1580 (1963).
80. Шемякин M. M., Овчинников Ю. А., Иванов В. Т. и Кирюшкин А. А., Tetrahedron Letters, 28, 1927 (1963).
81. Шляпникоё С. В., Карпейский M. Я., Литвин Е. Ф., Биохимия, 28, 664; Chem. Abstr., 59, 14278 с (1963).
82. Simmons Н. Е., Wiley D. W., J. Am. Chem. Soc., 82, 2288 (1960).
83. Stalling D. L., Gille G., Gehrke C. W., Anal. Biochem., 18, 118 (1967).
84. Steglich W., Austel V., Chem. Ber., 100, 547 (1967).
85. Stenhagen E., Z. Anal. Chem., 181, 462 (1961).
86. Stevenson J. M., Luck J. W., J. Biol. Chem., 236, 715 (1961).
87. Tanner H., Dissertation, Techn. Hochschule (Technical University), München, 1963.
88. Teuwissen B., Lenain C., Dorlet C., Leonis J., J. Pharm. Belg., 88 (1963).
89. Ulehla J., Sbornik sceskoslov. Akad. Zemedelske ved., Zivocisna vybora, 5 567, (1960); Chem. Abstr., 55, 5542 h. (1961).
90. Van den Heuvel W. J. A., Anal. Chem., 35, 526 (1963).
91. Van den Heuvel W. J. A., Horning E. C., Anal. Cnem., 36 (1964).
92. Virtanen A. I., Rautanen N., Suomen Kemistilehti, 19B, 56; Chem. Abstr., 41, 5566 (1946, 1947).
93. Витт С. В., Сопоровская M. Б., Беликов В. H., Изв. АН Латв. ССР, хим. серия, 525 (1963).
94. Voss W., Blanke Е., Liebigs Ann. Chem., 485, 258 (1931).
95. Wagner J., Rausch G., Z. Anal. Chem., 194, 350 (1963).
96. Wagner J., Winkler G., Z. Anal. Chem., 183, 1 (1961).
97. Watson J. Th., Biemann K., Anal. Chem., 36, 1135 (1964).
98. Weinstein B., Fenschau A. H., J. Chromatogr., 15, 149 (1964).
99. Weygand F., Diskussionsbeitrag auf der GDCh-Hauptversammlung Berlin anläßlich eines Vortrags von F. Bayer am 9 Oktober (1957).
100. Weygand F., Z. Anal. Chem., 205, 406 (1964).
101. Weygand F., Z. Anal. Chem., 205, 413 (1964).
102. Weygand F., Burger K., Engelhardt K., Chem. Ber., 99, 1461 (1966).
103. Weygand F., Csendes E., Angew. Chem., 64, 136 (1952).
104. Weygand F., Eberhardt G., Angew. Chem., 64, 458 (1952).
105. Weygand F., Eichner K., неопубликованные данные.
106. Weygand F., Fritz H., Chem. Ber., 98, 72 (1965).
107. Weygand F., Geiger R., Chem. Ber., 89, 647 (1956).
108. Weygand F., Geiger R., Swodenk W., Angew. Chem., 68, 307 (1956).
109. Weygand F., Geiger R., Chem. Ber., 92, 2099 (1959).
110. Weygand F., Glöckner U., Chem. Ber., 89, 653 (1956).
111. Weygand F., Raelicke J., неопубликованные данные.
112. Weygand F., Klinke P., Eigen I., Chem. Ber., 90, 1896 (1957).
113. Weygand F., Klipping G., Palm D., Chem. Ber., 93, 2619 (1960).
114. Weygand F., Kolb B., Kirchner P., Z. Anal. Chem., 181, 396 (1961).
115. Weygand F., Kolb B., Prox A., Tilak M. A., Tomida I., Hoppe-Seylers, Z. Physiol. Chem., 322, 38 (1960).
116. Weygand F., König W., Prox A., Burger K., Chem. Ber., 99, 1443 (1966),
117. Weygand F., Leising E., Chem. Ber., 87, 248 (1954).
118. Weygand F., Nintz E., неопубликованные данные.
119. Weygand F., Prox А., неопубликованные данные.
120. Weygand F., Prox A., Fessel H.-H., Sun К. K., Z. Naturforsch, 20b, 1169 (1965).
121. Weygand F., Prox A., Jorgensen E. C., Axen R., Kirchner P., Z. Naturforsch., 186, 93 (1963).
122. Weygand F., Prox А., König W., неопубликованные данные.
123. Weygand F., Prox A., König W., Fessel H.-H., Angew. Chem., 75, 724(1963).
124. Weygand F., Prox A., Schnidhammer L., König W., Angew. Chem., 75, 282 (1963).
125. Weygand F., Reiher M., Chem. Ber., 88, 27 (1955).
126. Weygand F., Rinno H., Chem. Ber., 92, 517 (1959).
127. Weygand F., Steglich W., Tanner H., Liebigs Ann. Chem., 658, 128 (1962).
128. Winter L. N., Albro P. W., J. Gas-Chromatogr., 2, 1 (1964).
129. Youngs C. G., Anal. Chem., 31, 1019 (1959).
130. Zlatkis A., Oro J. F., Kimbal A. P., Anal. Chem., 32, 162 (1960).
131. Zomzely G., Marco G., Emery E., Anal. Chem., 34, 1414 (1962).