БИОХИМИЯ УЧЕБНИК ДЛЯ ВУЗОВ - Е. С. Северина - 2004
РАЗДЕЛ 10. ОБМЕН НУКЛЕОТИДОВ
IV. Нарушения обмена пуриновых нуклеотидов
Ураты значительно более растворимы, чем мочевая кислота: так, в моче с pH 5,0, когда мочевая кислота не диссоциирована, её растворимость в 10 раз меньше, чем в моче с pH 7,0, при котором основная часть мочевой кислоты представлена солями. Реакция мочи зависит от состава пищи, но, как правило, она слабокислая, поэтому большинство камней в мочевыводящей системе — кристаллы мочевой кислоты.
А. Гиперурикемия и подагра
Когда в плазме крови концентрация мочевой кислоты превышает норму, то возникает гиперурикемия. Вследствие гиперурикемии может развиться подагра — заболевание, при котором кристаллы мочевой кислоты и уратов откладываются в суставных хрящах, синовиальной оболочке, подкожной клетчатке с образованием подагрических узлов, или тофусов. К характерным признакам подагры относят повторяющиеся приступы острого воспаления суставов (чаще всего мелких) — так называемого острого подагрического артрита. Заболевание может прогрессировать в хронический подагрический артрит.
Поскольку лейкоциты фагоцитируют кристаллы уратов, то причиной воспаления является разрушение лизосомальных мембран лейкоцитов кристаллами мочевой кислоты. Освободившиеся лизосомальные ферменты выходят в цитозоль и разрушают клетки, а продукты клеточного катаболизма вызывают воспаление.
Общий фонд сывороточных уратов в норме составляет ~ 1,2 г у мужчин и 0,6 г у женщин. При подагре без образования тофусов (т. е. подагрических узлов, в которых накапливаются ураты натрия и мочевая кислота) количество уратов возрастает до 2 — 4 г, а у пациентов с тяжёлой формой болезни, сопровождающейся ростом тофусов, может достигать 30 г.
Подагра — распространённое заболевание, в разных странах ею страдают от 0,3 до 1,7% населения. А поскольку сывороточный фонд уратов у мужчин в 2 раза больше, чем у женщин, то они и болеют в 20 раз чаще, чем женщины.
Как правило, подагра генетически детерминирована и носит семейный характер. Она вызвана нарушениями в работе ФРДФ синтетазы или ферментов «запасного» пути: гипоксантин- гуанин- или аденинфосфорибозилтрансфераз.
К другим характерным проявлениям подагры относят нефропатию, при которой наблюдают образование уратных камней в мочевыводящих путях.
Полиморфные варианты ФРДФ синтетазы
Активность ФРДФ синтетазы, катализирующей образование ФРДФ, строго контролируется пуриновыми нуклеотидами. Мутации в гене ФРДФ синтетазы привели к появлению полиморфных вариантов фермента, которые характеризуются аномальным ответом на обычные регуляторные факторы: концентрацию рибозо-5-фосфата и пуриннуклеотидов. Как правило, наблюдается суперактивация фермента. Пуриновые нуклеотиды синтезируются со скоростью, почти независимой от нужд клетки. Это вызывает ингибирование запасных «путей спасения», усиление катаболизма избыточного количества нуклеотидов, повышение продукции мочевой кислоты, гиперурикемию и подагру (табл. 10-1).
Таблица 10-1. Гиперурикемия, вызванная дефектами в работе ферментов обмена пуриннуклеотидов
Дефектный фермент |
Характер дефекта |
Клинические проявления |
Заболевание |
ФРДФ синтетаза |
Суперактивация и ↑ Vmах Устойчивость к ретроингибированию Снижение Кm для рибозо- 5-фосфата |
Гиперурикемия, повышенная экскреция уратов с мочой, подагрический артрит |
Подагра |
Гипоксантин-гуанинфосфори- бозилтрансфераза |
Частичная потеря активности Полная потеря активности |
Те же Гиперурикемия, нефропатия, артрит, неврологические и психические отклонения |
Подагра Синдром Лёша-Нихена |
Аденинфосфорибозилтрансфераза |
Полная потеря активности |
Образование камней 2,8- дигидроксиаденина |
Почечнокаменная болезнь |
Примерно у 40% больных одной из форм гликогеноза — болезнью Гирке (недостаточностью глюкозо-6-фосфатазы) сопутствующей патологией является подагра. Снижение способности печени секретировать глюкозу в кровь увеличивает использование глюкозо-6-фосфата в пентозофосфатном пути. Образуются большие количества рибозо-5-фосфата, которые могут стимулировать избыточный синтез, а, следовательно, и катаболизм пуриновых нуклеотидов.
Б. Недостаточность ферментов ‹запасных путей› синтеза пуриновых нуклеотидов. Синдром Леша-Нихена
В ряде случаев причиной гиперурикемии, избыточной экскреции пуринов с мочой и подагры являются нарушения в работе ферментов «пути спасения» пуриновых оснований (табл. 10-1). Гипоксантин-гуанин фосфорибозилтрансфераза катализирует реакцию превращения гуанина и гипоксантина в соответствующие нуклеотиды (рис. 10-7). Обнаружены полиморфные варианты гипоксантин-гуанинфосфорибозилтрансферазы со сниженной ферментативной активностью, что:
✵ уменьшает повторное использование пуриновых оснований, и они превращаются в мочевую кислоту;
✵ увеличивает синтез пуриновых нуклеотидов de novo из-за слабого использования ФРДФ в реакциях реутилизации и увеличения его концентрации в клетке. Адениловые и гуаниловые нуклеотиды образуются в количествах, превышающих потребности клеток, а это способствует усилению их катаболизма.
Синдром Лёша-Нихена — тяжёлая форма гиперурикемии, которая наследуется как рецессивный признак, сцепленный с Х-хромосомой, и проявляется только у мальчиков.
Болезнь вызвана полным отсутствием активности гипоксантин-гуанинфосфорибозилтрансферазы и сопровождается гиперурикемией с содержанием мочевой кислоты от 9 до 12 мг/дл, что превышает растворимость уратов при нормальном pH плазмы. Экскреция мочевой кислоты у больных с синдромом Лёша-Нихена превышает 600 мг/сут и требует для выведения этого количества продукта не менее 2700 мл мочи.
У детей с данной патологией в раннем возрасте появляются тофусы, уратные камни в мочевыводящих путях и серьёзные неврологические отклонения, сопровождающиеся нарушением речи, церебральными параличами, снижением интеллекта, склонностью к нанесению себе увечий (укусы губ, языка, пальцев).
В первые месяцы жизни неврологические расстройства не обнаруживаются, но на пелёнках отмечают розовые и оранжевые пятна, вызванные присутствием в моче кристаллов мочевой кислоты. При отсутствии лечения больные погибают в возрасте до 10 лет из-за нарушения функции почек.
Полная потеря активности аденинфосфорибозилтрансферазы не столь драматична, как отсутствие гипоксантин-гуанинфосфорибозилтрансферазы, однако и в этом случае нарушение повторного использования аденина вызывает гиперурикемию и почечнокаменную болезнь, при которой наблюдается образование кристаллов 2,8-дигидроксиаденина.
В. Лечение гиперурикемии
Основным препаратом, используемым для лечения гиперурикемии, является аллопуринол — структурный аналог гипоксантина (рис. 10-11).
Рис. 10-11. Строение аллопуринола и гипоксантина.
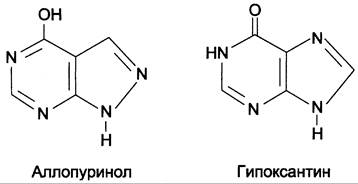
Аллопуринол оказывает двоякое действие на обмен пуриновых нуклеотидов:
✵ ингибирует ксантиноксидазу и останавливает катаболизм пуринов на стадии образования гипоксантина, растворимость которого почти в 10 раз выше, чем мочевой кислоты. Действие препарата на фермент объясняется тем, что сначала он, подобно гипоксантину, окисляется в гидроксипуринол, но при этом остаётся прочно связанным с активным центром фермента, вызывая его инактивацию;
✵ с другой стороны, будучи псевдосубстратом, аллопуринол может превращаться в нуклеотид по «запасному» пути и ингибировать ФРДФ синтетазу и амидофосфорибозил- трансферазу, вызывая торможение синтеза пуринов de novo.
При лечении аллопуринолом детей с синдромом Лёша-Нихена удаётся предотвратить развитие патологических изменений в суставах и почках, вызванных гиперпродукцией мочевой кислоты, но препарат не излечивает аномалии в поведении, неврологические и психические расстройства.
Г. Гипоурикемия
Гипоурикемия и возросшая экскреция гипоксантина и ксантина может быть следствием недостаточности ксантиноксидазы, вызванной нарушениями в структуре гена этого фермента, либо результатом повреждения печени.