БИОХИМИЯ УЧЕБНИК ДЛЯ ВУЗОВ - Е. С. Северина - 2004
РАЗДЕЛ 14. БИОХИМИЯ КРОВИ
III. Свертывающая система крови
При повреждении кровеносного сосуда инициируется каскад реакций, в результате которого образуется сгусток крови — тромб, предотвращающий кровотечение. Основную роль в свёртывании (коагуляции) крови играют тромбоциты и ряд белков плазмы крови.
В остановке кровотечения различают 3 этапа. На первом этапе происходит сокращение кровеносного сосуда. Затем к месту повреждения прикрепляются тромбоциты, которые, наслаиваясь друг на друга, образуют тромбоцитарную пробку (белый тромб). Белый тромб является непрочным и может закупорить только небольшой кровеносный сосуд. На третьем этапе растворимый белок плазмы крови фибриноген превращается в нерастворимый белок фибрин, который откладывается между тромбоцитами, и формируется прочный фибриновый тромб. Такой тромб содержит эритроциты и поэтому называется красным тромбом.
Образованию фибринового тромба предшествует каскад протеолитических реакций, приводящий к активации фермента тромбина, который и превращает фибриноген в фибрин. Все белки, участвующие в свёртывании крови, называют факторами свёртывания. Они синтезируются в основном в печени и клетках крови в виде неактивных предшественников, обозначаются римскими цифрами, но имеют и тривиальные названия (табл. 14-1). Большинство этих белков активируется в каскаде ферментативных реакций свёртывания крови. Активные формы этих белков обозначают такими же римскими цифрами, но с добавлением буквы «а».
Таблица 14-1. Основные функции и содержание в плазме крови факторов свертывания крови
Фактор |
Тривиальное название |
Содержание в плазме крови, г/л |
Функции |
1 |
2 |
3 |
4 |
I |
Фибриноген |
2-4 |
Растворимый белок-предшественник фибрина |
Iа |
Фибрин |
Образует фибриновый гель |
|
II |
Протромбин |
0,1 |
Профермент* |
IIа |
Тромбин |
Протеаза, превращающая фибриноген в фибрин и активирующая факторы V, VII, VIII, XIII, С |
|
III |
Тканевый фактор |
Белок-активатор мембранного комплекса VIIa-ТФ- Са2+ |
|
IV |
Са2+ |
0,9-1,2 ммоль/л |
Опосредует взаимодействие ферментов прокоагулянтного пути с фосфатидилсерином |
V |
Проакцелерин |
0,01 |
Предшественник белка-активатора мембранного комплекса Xa-Va-Ca2+ |
Va |
Акцелерин |
Белок-активатор мембранного комплекса Xa-Va- Са2+ |
|
VII |
Проконвертин |
0,005 |
Профермент* |
Vlla |
Конвертин |
Протеаза*, активирующая факторы X и IX |
|
VIII |
Неактивный антигемофильный фактор А (неактивный антигемофильный глобулин) |
0,01-0,02 |
Предшественник белка-активатора мембранного комплекса IXa-VIIIa-Ca2+ |
VIIla |
Активный антигемофильный фактор А (активный антигемофильный глобулин) |
Белок-активатор мембранного комплекса IXa-VIIIa-Ca2+ |
|
IX |
Неактивный антигемофильный фактор В (неактивный фактор Кристмаса) |
0,003 |
Профермент* |
IXa |
Активный антигемофильный фактор В (активный фактор Кристмаса) |
Протеаза*, активирующая фактор X |
|
X |
Неактивный фактор Стюарта-Прауэра |
0,01 |
Профермент* |
Xa |
Активный фактор Стюарта-Прауэра |
Протеаза*, активирующая фактор II |
|
XI |
Неактивный плазменный предшественник тромбопластина |
0,005 |
Профермент контактного пути свёртывания крови |
ХIа |
Активный плазменный предшественник тромбопластина |
Протеаза, активирующая фактор IX |
|
XII |
Неактивный фактор Хагемана |
0,03 |
Профермент контактного пути свёртывания крови |
ХIIа |
Активный фактор Хагемана |
Протеаза, активирующая фактор XI, прекалликреин, плазминоген |
|
XIII |
Неактивная трансглутамидаза (неактивный фибринста- билизирующий фактор) |
0,01-0,02 |
Профермент |
ХIIIа |
Активная трансглутамидаза (активный фибринстаби- лизирующий фактор) |
Катализирует образование амидных связей между молекулами фибрина-мономера, фибрином и фибронектином |
|
Прекалликреин Калликреин |
0,05 |
Профермент контактного пути свёртывания крови Протеаза, активирующая фактор XII, плазминоген |
|
ВМК |
0,06 |
Белок-активатор контактного пути свёртывания крови |
* Содержит остатки карбоксиглутаминовой кислоты, необходимые для образования мембранных ферментных комплексов прокоагулянтного пути свёртывания крови.
А. Образование фибринового тромба
Образование фибринового тромба начинается с превращения растворимого белка плазмы крови фибриногена в нерастворимый фибрин.
Фибриноген (фактор I) — гликопротеин с молекулярной массой 340 кД. Он синтезируется в печени и содержится в плазме крови в концентрации 8,02 — 12,9 мкмоль/л (2 — 4 г/л). Молекула фибриногена состоит из шести полипептидных цепей, которые связаны друг с другом дисульфидными связями. Состав полипептидных цепей молекулы фибриногена обозначают Аα2, Вβ2, y2. Заглавные буквы соответствуют тем участкам, которые отщепляются под действием тромбина при превращении фибриногена в фибрин. Фрагменты А в цепях Аα и В в цепях Вβ содержат большое количество остатков аспартата и глутамата. Это создаёт сильный отри
цательный заряд на N-концах молекул фибриногена и препятствует их агрегации.
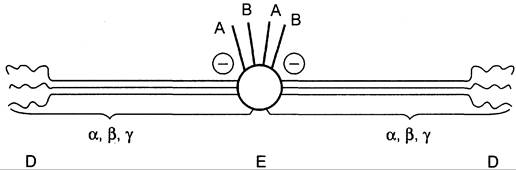
Молекула фибриногена состоит из трех глобулярных доменов, по одному на каждом конце молекулы (домены Д) и один в середине (домен Е). Домены отделены друг от друга участками полипептидных цепей, имеющими стержнеобразную конфигурацию. Из центрального домена Е выступают N-концевые фрагменты А и В цепей Аα и Вβ (рис. 14-8).
Рис. 14-8. Строение фибриногена. Фибриноген состоит из шести полипептидных цепей: Аα2, Вβ2 и y2. А, В — отрицательнозаряженные фрагменты, благодаря которым молекулы фибриногена не агрегируют. Д, Е — глобулярные домены молекулы фибриногена. Домены отделены участками полипептидных цепей, имеющими стержнеобразную конфигурацию. Из центрального глобулярного домена Е выступают N-концевые участки фрагментов А и В цепей Аα2 и Вβ2.
В образовании фибринового тромба можно выделить 4 этапа.
1. Превращение фибриногена в мономер фибрина.
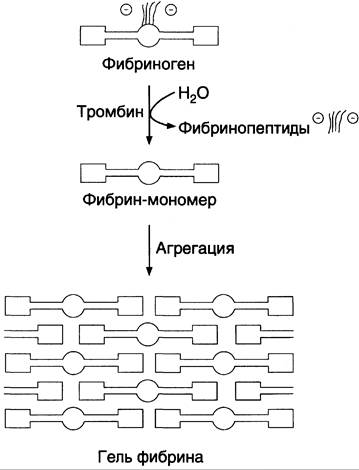
Сначала молекулы фибриногена освобождаются от отрицательно заряженных фрагментов А и В, в результате чего образуются мономеры фибрина. Превращение фибриногена (фактор I) в фибрин (фактор Iа) катализирует фермент тромбин (фактор IIа). В каждой молекуле фибриногена тромбин гидролизует четыре пептидные связи аргинил-глицил, две из которых соединяют фрагменты А с α-цепью, а две другие — В с β-цепью в Аα2- и Вβ2-цепях фибриногена. Мономер фибрина, образующийся из фибриногена, имеет состав (α, β, y)2.
2. Образование нерастворимого геля фибрина.
На втором этапе образуется нерастворимый полимерный фибриновый сгусток — гель фибрина. В результате превращения фибриногена в фибрин-мономер в домене Е открываются центры связывания с доменами D. Причём домен Е содержит центры агрегации, формирующиеся только после частичного протеолиза фибриногена под действием тромбина, а домен D является носителем постоянных центров агрегации. Первичная агрегация молекул фибрина происходит в результате взаимодействия центров связывания домена Е одной молекулы с комплементарными им участками на доменах D других молекул. Таким образом, между доменами молекул фибрина-мономера образуются нековалентные связи. При «самосборке» геля фибрина сначала образуются двунитчатые протофибриллы, в которых молекулы фибрина смещены друг относительно друга на 1/2 длины. После достижения протофибриллами определённой критической длины начинается их латеральная ассоциация, ведущая к образованию толстых фибриновых волокон (рис. 14-9). Образовавшийся гель фибрина непрочен, так как молекулы фибрина в нём связаны между собой нековалентными связями.
Рис. 14-9. Образование геля фибрина. Фибриноген, освобождаясь под действием тромбина от отрицательно заряженных фрагментов (фибринопептидов 2А и 2В), превращается в фибрин-мономер. В результате взаимодействия комплементарных участков Е- и D-доменов фибрина-мономера происходит сначала линейная, а затем латеральная полимеризация молекул с образованием геля фибрина.
3. Стабилизация геля фибрина.
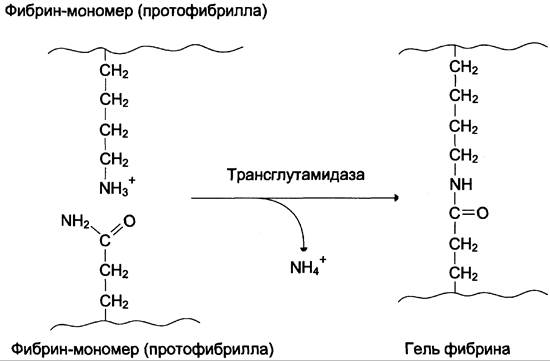
В результате образования амидных связей между остатками лизина одной молекулы фибрина и остатками глутамина другой молекулы гель фибрина стабилизируется. Реакцию трансамидирования катализирует фермент трансглутамидаза (фактор ХIIIа) (рис. 14-10). Фактор XIII активируется частичным протеолизом под действием тромбина.
Рис. 14-10. Образование амидной связи между молекулами фибрина.
Трансглутамидаза также образует амидные связи между фибрином и фибронектином — гликопротеином межклеточного матрикса и плазмы крови (см. раздел 15). Таким образом, тромб фиксируется в месте повреждения сосуда.
4. Ретракция фибринового сгустка.
Сжатие (ретракцию) геля обеспечивает актомиозин тромбоцитов — сократительный белок тромбостенин, обладающий АТФ-азной активностью. Тромбостенин участвует также в активации и агрегации тромбоцитов. Ретракция кровяного сгустка предупреждает полную закупорку сосудов, создавая возможность восстановления кровотока.
В механизме образования тромба есть три функционально разных этапа: прокоагулянтный путь, контактный путь и антикоагулянтная фаза, препятствующая распространению тромба.
Б. Прокоагулянтный путь вертывания крови
Для остановки кровотечения из капилляров и сосудов необходимо быстрое образование прочного тромба, препятствующего потере крови. Это достигается каскадом ферментативных реакций с механизмами усиления на многих этапах.
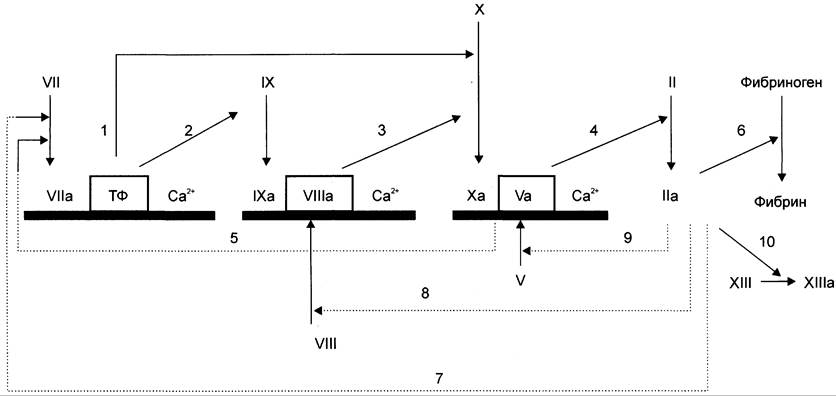
Прокоагулянтный путь занимает центральное место в свёртывании крови (рис. 14-11).
Рис. 14-11. Прокоагулянтный путь свёртывания крови. активация факторов свертывания крови; —> Рис. активация факторов свертывания крови по принципу положительной обратной связи;![]() мембранный фосфолипидный компонент ферментных комплексов. В рамку обведены белки-активаторы. 1,2 — фактор VIla мембранного комплекса Vlla-TФ-CA2+ активирует факторы IX и X; 3 — фактор IXa мембранного комплекса IXa-Vllla-Ca2+ активирует фактор X; 4, 5 — фактор Ха мембранного комплекса Xa-Va-Ca2+превращает протромбин (фактор II) в тромбин (фактор На) и активирует фактор VII; 6-10 — тромбин (фактор IIа) превращает нерастворимый фибриноген в растворимый фибрин, активирует факторы VIl, VIII, V и XIII.
мембранный фосфолипидный компонент ферментных комплексов. В рамку обведены белки-активаторы. 1,2 — фактор VIla мембранного комплекса Vlla-TФ-CA2+ активирует факторы IX и X; 3 — фактор IXa мембранного комплекса IXa-Vllla-Ca2+ активирует фактор X; 4, 5 — фактор Ха мембранного комплекса Xa-Va-Ca2+превращает протромбин (фактор II) в тромбин (фактор На) и активирует фактор VII; 6-10 — тромбин (фактор IIа) превращает нерастворимый фибриноген в растворимый фибрин, активирует факторы VIl, VIII, V и XIII.
В циркулирующей крови содержатся проферменты протеолитических ферментов: фактор II (протромбин), фактор VII (проконвертин), фактор IX (Кристмаса), фактор X (Стюарта). Находящиеся в крови факторы Va (акцелерин) и VIlla (антигемофильный фактор), а также мембранный белок — тканевый фактор (ТФ, фактор III) являются белками-активаторами этих ферментов (табл. 14-1).
При повреждении сосуда «включается» каскадный механизм активации ферментов с последовательным образованием трёх связанных с фосфолипидами клеточной мембраны ферментных комплексов. Каждый комплекс состоит из протеолитического фермента, белка- активатора и ионов Са2+: VIIa-TФ-Ca2+, IХа- VIIIа-Са2+ (теназа), Ха-Vа-Са2+(протромбиназа) (рис. 14-12). Комплекс Ха-Vа-Са2+ (протромбиназный комплекс) активирует протромбин (фактор II). Каскад ферментативных реакций завершается образованием мономеров фибрина и последующим формированием тромба.
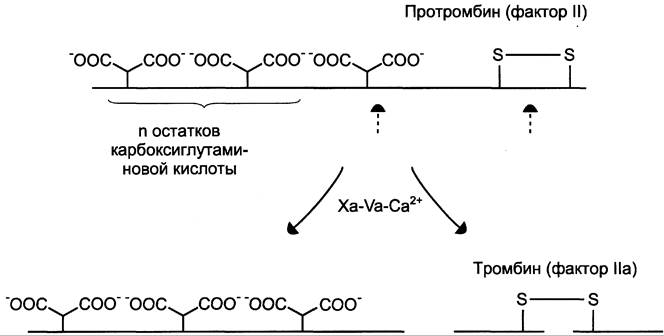
Рис. 14-12. Протеолитическая активация протромбина фактором Ха протромбиназного комплекса.![]() остатки карбоксиглутаминовой кислоты; штрих-стрелки указывают положение гидролизуемых в молекуле протромбина пептидных связей. Молекула протромбина состоит из одной полипептидной цепи, а образующийся в результате частичного протеолиза протромбина тромбин состоит из двух полипептидных цепей, связанных между собой одной дисульфидной связью.
остатки карбоксиглутаминовой кислоты; штрих-стрелки указывают положение гидролизуемых в молекуле протромбина пептидных связей. Молекула протромбина состоит из одной полипептидной цепи, а образующийся в результате частичного протеолиза протромбина тромбин состоит из двух полипептидных цепей, связанных между собой одной дисульфидной связью.
В активации ферментов каскада выделяют три основных механизма: частичный протеолиз, взаимодействие с белками-активаторами и взаимодействие с модифицированными клеточными мембранами.
Активация частичным протеолизом. Все ферменты прокоагулянтного пути являются сериновыми протеазами, синтезируются в печени в виде неактивных проферментов и в такой форме циркулируют в крови. В процессе реализации тромбогенного сигнала проферменты (факторы VII, IX, X и II) частичным протеолизом превращаются в активные ферменты.
Тромбин (фактор IIа) — гликопротеин с молекулярной массой 39 кД. Он образуется в крови из неактивного предшественника протромбина. Протромбин синтезируется в печени, имеет молекулярную массу 70 кД и содержит остатки y-карбоксиглутаминовой кислоты. Концентрация этого белка в крови в норме составляет 0,1 г/л. Он фиксируется на мембранном ферментном комплексе Xa-Va-Ca2+, взаимодействуя, с одной стороны, остатками y-карбоксиглутамата с Са2+, а с другой — непосредственно с белком-активатором Va. Таким образом, создаются наилучшие стерические условия для протекания ферментативной реакции. Фактор Ха гидролизует две пептидные связи в молекуле протромбина. В результате этого образуется молекула тромбина, состоящая из двух цепей — лёгкой и тяжёлой, связанных между собой одной дисульфидной связью (рис. 14-12). Молекула тромбина не содержит остатков y-карбоксиглутамата и освобождается из протромбиназного комплекса. Тромбин частичным протеолизом превращает фибриноген в фибрин и активирует факторы VII, VIII, V, XIII.
Тромбин выполняет ряд важных физиологических функций: является ферментом прокоагулянтного и контактного путей свёртывания крови, инициирует реакции антикоагулянтной фазы, вызывает агрегацию тромбоцитов и оказывает митогенное действие, участвуя в пролиферации и репарации клеток.
Частичным протеолизом активируются также факторы V и VIII, превращаясь, соответственно, в факторы Va и VIlla. В результате активации этих факторов изменяется их конформация и повышается сродство к фосфолипидам мембран и ферментам, которые они активируют.
Взаимодействие белков-активаторов с протеолитическими ферментами. Тканевый фактор, фактор Va и фактор VIIla имеют центры связывания с фосфолипидами мембран и ферментами VIla, IXa и Ха, соответственно. При связывании с белками-активаторами в результате конформационных изменений активность этих ферментов повышается.
Тканевый фактор (фактор III) представляет собой комплекс, состоящий из белка и фосфатидилсерина. Белковая часть тканевого фактора (апопротеин III) экспонирована на поверхности многих клеток (мозга, лёгких, печени, селезёнки и др.) и связана с фосфатидилсерином плазматических мембран. Однако появление апопротеина III на поверхности клеток, соприкасающихся с кровью (эндотелиальных и моноцитов), происходит только при определённых условиях: при повреждении сосуда и/или нарушении нормальной асимметрии их плазматических мембран. Тканевый фактор в протеолитической активации не нуждается.
Фактор V и фактор VIII — доменные белки, циркулирующие в крови. Фактор V синтезируется в печени, а фактор VIII — эндотелиальными клетками. Оба фактора активируются частичным протеолизом под действием тромбина. Фактор VIII в плазме крови находится в комплексе с белком — фактором тромбоцитов фон Виллебранда. Фактор фон Виллебранда в этом комплексе стабилизирует фактор VIII, препятствуя его разрушению протеолитическим ферментом антикоагулянтной фазы фактором Са.
Взаимодействие ферментных комплексов с клеточными мембранами происходит с участием ионов Са2+. Все проферменты прокоагулянтного пути (II, VII, IX, X) содержат остатки у-карбоксиглутаминовой кислоты, образующиеся в результате посттрансляционой модификации этих белков в ЭР гепатоцитов.
Остатки y-карбоксиглутаминовой кислоты в факторах VIla, IXa и Ха обеспечивают взаимодействие этих ферментов посредством Са2+ с отрицательно заряженными фосфолипидами клеточных мембран. В отсутствие ионов Са2+ кровь не свёртывается.
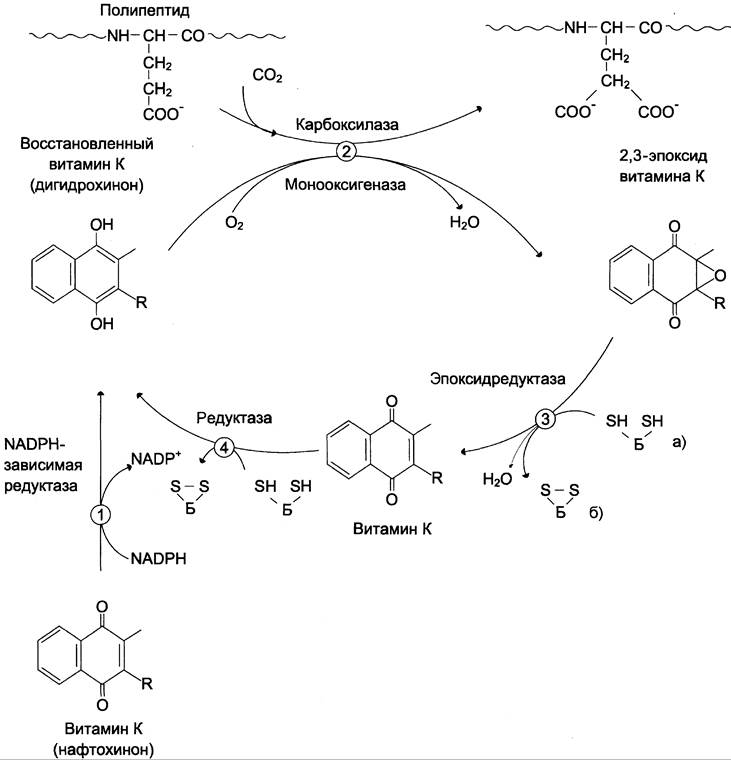
Роль витамина К в карбоксилировании остатков глутаминовой кислоты в проферментах прокоагулянтного пути свёртывания крови. Карбоксилирование остатков глутаминовой кислоты в проферментах прокоагулянтного пути катализирует карбоксилаза, коферментом которой служит восстановленная форма витамина К (нафтохинона) — дигидрохинон витамина К (см. раздел 3).
Поступивший в организм витамин К (нафтохинон) восстанавливается в печени NАDРН-зависимой витамин К редуктазой с образованием дигидрохинона витамина К. В ходе реакции карбоксилирования остатков глутаминовой кислоты в проферментах прокоагулянтного пути дигидрохинон окисляется и эпоксидируется с образованием 2,3-эпоксида витамина К. Регенерация эпоксида в дигидрохинон витамина К происходит следующим образом: сначала 2,3- эпоксид витамина К восстанавливается в витамин К тиолзависимой эпоксидредуктазой, коферментом которой является белок, подобный тиоредоксину. Затем образующийся в этой реакции витамин К восстанавливается ферментом витамин К тиолзависимой редуктазой в ди- гидрохинон витамина К. Донором водорода в этой реакции, так же, как и в предыдущей, служит тиоредоксинподобный белок (рис. 14-13).
Рис. 14-13. Роль витамина К в посттрансляционном карбоксилировании глутаминовой кислоты. 1 — восстановление экзогенного витамина К NADPH-зависимой редуктазой; 2 — y-карбоксилирование остатков глутаминовой кислоты в факторах II, VII, IX, X, протеине С витамин К зависимой карбоксилазой сопровождается окислением дигидрохинона с образованием 2,3- эпоксида витамина К; 3 — восстановление 2,3-эпоксида тиолзависимой витамин К редуктазой; 4 — восстановление витамина К тиолзависимой витамин К редуктазой; а) и б) — восстановленная и окисленная формы тиоредоксинподобного белка.
Недостаточность витамина К приводит к нарушению карбоксилирования проферментов прокоагулянтного пути и сопровождается кровоточивостью, подкожными и внутренними кровоизлияниями.
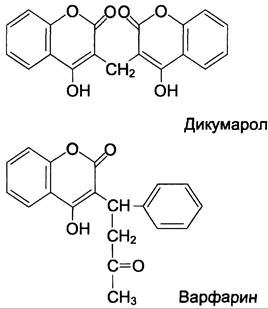
Структурные аналоги витамина К дикумарол и варфарин ингибируют тиолзависимые ферменты витамин К 2,3-эпоксидредуктазу и витамин К редуктазу, вызывая торможение свёртывания крови (рис. 14-14). Эти препараты применяют в клинической практике для предупреждения тромбозов.
Рис. 14-14. Структурные аналоги витамина К дикумарол и варфарин.
Инициация каскада реакций прокоагулянтного пути. Ферментные мембранные комплексы прокоагулянтного пути образуются только при наличии на внешней поверхности плазматической мембраны клеток тканевого фактора и отрицательно заряженных фосфолипидов. Поперечная асимметрия плазматических мембран, в частности, определяется преобладанием в наружном слое нейтральных фосфолипидов (фосфатидил- холина и сфингомиелина), а во внутреннем — отрицательно заряженных (фосфатидилинозитол- бисфосфата и фосфатидилсерина) (см. раздел 5). Специальная ферментная система обеспечивает трансмембранный перенос и такое распределение фосфолипидов в клеточных мембранах, при котором в норме внешняя поверхность плазматических мембран клеток не заряжена (см. раздел 5).
При нарушении поперечной асимметрии мембран тромбоцитов и эндотелиальных клеток на их поверхности формируются отрицательно заряженные (тромбогенные) участки и экспонируется апопротеин III тканевого фактора. Такие нарушения могут возникнуть при физической травме. В этом случае тканевый фактор и внутренняя поверхность клеточной мембраны становятся доступными для плазменных факторов прокоагулянтного пути. Кроме того, взаимодействие сигнальных молекул, вызывающих тромбогенез, с рецепторами эндотелиальных клеток и тромбоцитов активирует Са2+-зависимые регуляторные системы (см. раздел 5). В конечном итоге это приводит к повышению содержания в цитоплазме Са2+, который ингибирует АТФ-зависимую аминофосфолипидтранслоказу. Этот фермент играет важную роль в сохранении поперечной асимметрии мембран, так как переносит фосфа- тидилсерин из внешнего липидного слоя во внутренний. Снижение активности аминофосфолипидтранслоказы приводит к увеличению содержания во внешнем слое клеточной мембраны фосфатидилсерина и образованию отрицательно заряженных участков, необходимых для формирования мембранных ферментных комплексов. Кроме того, в результате такого нарушения структуры плазматической мембраны на её внешней поверхности экспонируется тканевый фактор и формируется первый ферментный комплекс прокоагулянтного пути свёртывания крови VII-ТФ-Са2+.
Активация ферментов каждого комплекса — результат взаимодействия всех его компонентов. Если факторы IX, X и II требуют активации, то фактор VII обладает невысокой протеолитической активностью. Фактор VII мембранного комплекса VII-ТФ-Са2+ частичным протеолизом активирует факторы IX и X. Активные факторы 1Ха и Ха включаются в образование мембранных комплексов IXa-VIIIa-Ca2+ и Ха- Va-Ca2+. При этом фактор Ха протеолитически активирует фактор V, а протромбиназный комплекс не только превращает протромбин в тромбин, но и активирует фактор VII, протеолитическая активность которого в комплексе Vlla- Тф-Са2+ в 10 000 раз выше, чем в комплексе VII-Тф-Са2+.
Образовавшийся в результате каскада реакций тромбин катализирует реакции частичного протеолиза фибриногена, фактора XIII и по принципу положительной обратной связи протеолитически активирует факторы V, VII и VIII.
В процессе свёртывания действуют 2 механизма усиления сигнала: каскад реакций, в котором каждое ферментативное звено обеспечивает усиление сигнала, и положительные обратные связи.
В. Контактный путь свертывания крови
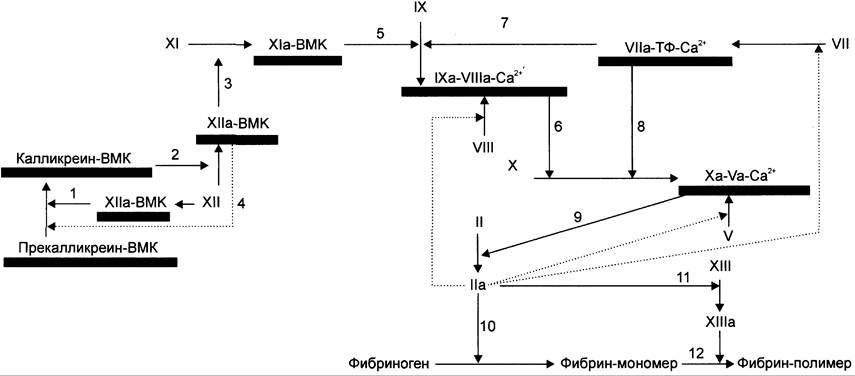
Контактный путь свёртывания крови начинается с взаимодействия профермента фактора XII с повреждённой эндотелиальной поверхностью сосудистой стенки. Такое взаимодействие приводит к активации фактора XII и инициирует образование мембранных ферментных комплексов контактной фазы свёртывания. Они содержат ферменты калликреин, факторы ХIа (плазменный предшественник тромбопластина) и ХIIа (фактор Хагемана), а также белок-активатор — высокомолекулярный кининоген (ВМК) (рис. 14-15).
Рис. 14-15. Схема прокоагулянтного (внешнего) и контактного (внутреннего) путей свёртывания крови. Обозначения: ВМК — высокомолекулярный кининоген; ТФ — тканевый фактор; —> — активация факторов свёртывания крови; активация факторов свёртывания по принципу положительной обратной связи;![]() — мембранный фосфолипидный компонент ферментных комплексов. Все ферменты мембранных комплексов свертывающей системы крови являются протеазами и активируются частичным протеолизом. 1 — активированный в результате контакта с субэндотелием фактор XII превращает прекалликреин в калликреин; 2 — калликреин мембранного комплекса калликреин-ВМК активирует фактор XII; 3 — фактор ХIIа активирует фактор XI; 4 — активированный частичным протеолизом фактор ХIIа превращает прекалликреин в калликреин по принципу положительной обратной связи; 5 — фактор Xla мембранного комплекса Xla-BMK активирует фактор IX; 6 — фактор IXa мембранного комплекса IXa-Vllla-Ca2+ активирует фактор X; 7, 8 — фактор VIla мембранного комплекса VIla- Тф-Са2+ активирует факторы IX и X; 9 — фактор Ха протромбиназного комплекса активирует фактор II; 10, 11 — тромбин (фактор II) превращает фибриноген в фибрин и активирует фактор XIII; 12 — фактор ХIIIа катализирует образование амидных связей в геле фибрина.
— мембранный фосфолипидный компонент ферментных комплексов. Все ферменты мембранных комплексов свертывающей системы крови являются протеазами и активируются частичным протеолизом. 1 — активированный в результате контакта с субэндотелием фактор XII превращает прекалликреин в калликреин; 2 — калликреин мембранного комплекса калликреин-ВМК активирует фактор XII; 3 — фактор ХIIа активирует фактор XI; 4 — активированный частичным протеолизом фактор ХIIа превращает прекалликреин в калликреин по принципу положительной обратной связи; 5 — фактор Xla мембранного комплекса Xla-BMK активирует фактор IX; 6 — фактор IXa мембранного комплекса IXa-Vllla-Ca2+ активирует фактор X; 7, 8 — фактор VIla мембранного комплекса VIla- Тф-Са2+ активирует факторы IX и X; 9 — фактор Ха протромбиназного комплекса активирует фактор II; 10, 11 — тромбин (фактор II) превращает фибриноген в фибрин и активирует фактор XIII; 12 — фактор ХIIIа катализирует образование амидных связей в геле фибрина.
Фактор XII — профермент, циркулирующий в крови. Он последовательно активируется двумя способами: сначала в результате изменения конформации при взаимодействии с отрицательно заряженной поверхностью повреждённого эндотелия, затем частичным протеолизом мембранным комплексом калликреин-ВМК.
Высокомолекулярный кининоген — белок-активатор в ферментных мембранных комплексах ХIIа-ВМК, ХIа-ВМК и калликреин-ВМК. ВМК — гликопротеин плазмы крови, который синтезируется в печени и имеет молекулярную массу 120 кД. Он опосредует взаимодействие протеолитических ферментов контактной фазы свёртывания крови с коллагеном субэндотелия и, кроме того, является компонентом калликреин-кининовой системы.
Калликреин — сериновая протеаза, субстратами которой являются, кроме фактора XII, белки плазмы крови плазминоген (профермент, участвующий в растворении фибрина) и кининогены с низкой (69 кД) и высокой (120 кД) молекулярной массой. При частичном протеолизе кининогенов образуются регуляторные пептиды кинины. В частности, мощный вазодилятатор брадикинин повышает проницаемость сосудов и вызывает разрушение клеточных мембран эндотелия.
В результате контакта фактора XII с субэндотелием сосудов он активируется. Активный фактор ХIIа в комплексе с ВМК протеолитически превращает прекалликреин, связанный с мембраной посредством ВМК, в калликреин. Мембранный комплекс калликреин-ВМК по принципу положительной обратной связи частичным протеолизом активирует фактор XII. При этом фактор XII приобретает максимальную ферментативную активность и по принципу положительной обратной связи активирует связанный с ВМК прекалликреин. Кроме того, образовавшийся в результате частичного протеолиза фактор ХПа протеолитически активирует фактор XI, а фактор ХIа в составе ферментного комплекса XIa-ВМК активирует фактор IX. Фактор IХа мембранного комплекса IХа- VIIIa-Ca2+активирует фактор X, который в составе протромбиназного комплекса активирует протромбин.
Каскад реакций, ведущий к образованию тромбина, может реализоваться двумя путями — прокоагулянтным (внешним) и контактным (внутренним) (рис. 14-15). Для инициации реакций внешнего пути необходимо появление тканевого фактора на внешней поверхности плазматической мембраны клеток, соприкасающихся с кровью. Внутренний путь начинается с активации фактора XII при его контакте с повреждённой поверхностью эндотелия сосудов и взаимной активации ферментов прекалликреина и фактора XII.
Таким образом, в прокоагулянтном и контактном путях свёртывания крови последовательное образование мембранных ферментных комплексов приводит к активации фактора X и образованию протромбиназы. Этапы, одинаковые для обоих путей свёртывания крови, называют общим путём свёртывания крови. В настоящее время понятие о внутреннем и внешнем путях свёртывания считают достаточно условным, так как стало ясно, что комплекс VIIа-ТФ-Са2+ более эффективно активирует фактор IX, чем фактор X, а фактор VII активируется фактором IХа, хотя и значительно медленнее по сравнению с активацией фактором Ха. Следовательно, можно полагать, что каскад реакций свёртывания крови идёт преимущественно в линейной последовательности, а не по двум относительно независимым путям. Контактный путь, очевидно, не является абсолютно необходимым для инициации свёртывания; по-видимому, он служит для сопряжения системы свёртывания крови с различными регуляторными системами организма, например, калликреин-кининовой и системой ферментов фибринолиза, растворяющей тромб.
Кровь здорового человека in vitro свёртывается за 5 — 10 мин. При этом образование протромбиназного комплекса занимает 5 — 8 мин, активация протромбина — 2 — 5 с и превращение фибриногена в фибрин — 2 — 5 с.
Снижение свёртываемости крови. При снижении свёртываемости крови наблюдают заболевания, сопровождающиеся повторяющимися кровотечениями. Гемофилии — наследственные болезни, характеризующиеся повышенной кровоточивостью. Причиной этих кровотечений (спонтанных или вызванных травмой) является наследственная недостаточность белков свёртывающей системы крови.
Гемофилия А (классическая гемофилия) обусловлена мутацией гена фактора VIII, локализованного в X хромосоме. Классическая гемофилия составляет 80% всех случаев заболевания гемофилией. Гемофилия В встречается реже и обусловлена генетическим дефектом фактора IX.
Дефект гена фактора VIII проявляется как рецессивный признак, поэтому этой формой гемофилии болеют только мужчины. Это заболевание сопровождается подкожными, внутримышечными и внутрисуставными кровоизлияниями, иногда опасными для жизни. Дефект фактора VIII встречается примерно у одного из 10 000 новорождённых. Больных лечат препаратами, содержащими фактор VIII, получаемыми из донорской крови или методами генной инженерии.
Г. Противосвертывающая система крови
Физиологические ингибиторы свёртывания крови играют важную роль в поддержании гемостаза, так как они сохраняют кровь в жидком состоянии и препятствуют распространению тромба за пределы повреждённого участка сосуда.
Тромбин, образующийся в результате реакций прокоагулянтного и контактного путей свёртывания крови, вымывается током крови из тромба. Он может инактивироваться при взаимодействии с ингибиторами ферментов свёртывания крови или активировать антикоагулянтную фазу, тормозящую образование тромба.
Антикоагулянтная фаза. Свёртывание крови должно быть ограничено не только в пространстве, но и во времени. Антикоагулянтная фаза ограничивает время существования активных факторов в крови и инициируется самим тромбином. Следовательно, тромбин, с одной стороны, ускоряет свёртывание крови, являясь последним ферментом каскада реакций коагуляции, а с другой — тормозит его, вызывая образование ферментных комплексов антикоагулянтной фазы на неповреждённом эндотелии сосудов. Этот этап представляет собой короткий каскад реакций, в котором кроме тромбина участвуют белок-активатор тромбомодулин (Тм), витамин К-зависимая сериновая протеаза протеин С, белок-активатор S и факторы Va и VIlla (рис. 14-16).
Рис. 14-16. Антикоагулянтная фаза. Тм — тромбомодулин; С — протеин С; Са — активный протеин С; S — протеин S; жирные линии — мембранно-связанный комплекс. 1 — тромбин (На) образует мембранный комплекс с белком тромбомодулином (Тм); 2 — тромбин в составе мембранного комплекса IIа-Тм-Са2+ активирует протеин С; 3 — активированный протеин С в составе ферментного мембранного комплекса Са- S-Ca2+ гидролизует по 2 пептидные связи в факторах Va и VIlla и превращает их в неактивные пептиды.
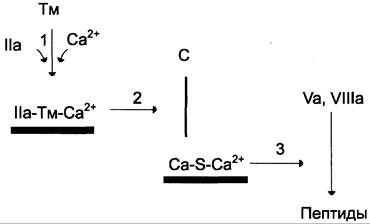
В каскаде реакций антикоагулянтной фазы последовательно образуются 2 мембранных комплекса IIа-Тм-Са2+ и Ca-S-Ca2+.
Тромбомодулин — интегральный белок мембран эндотелиальных клеток. Он не требует протеолитической активации и служит белком-активатором тромбина. Тромбин приобретает способность активировать протеин С только после взаимодействия с тромбомодулином, причём связанный с тромбомодулином тромбин не может превращать фибриноген в фибрин, не активирует фактор V и тромбоциты.
Протеин С — профермент, содержащий остатки y-карбоксиглутамата. Тромбин в мембранном комплексе IIа-Тм-Са2+ активирует частичным протеолизом протеин С. Активированный протеин С (Са) образует с белком-активатором S мембраносвязанный комплекс Ca-S-Ca2+. Са в составе этого комплекса гидролизует в факторах Va и Villa по две пептидные связи и инактивирует эти факторы. Под действием комплекса Ca-S-Ca2+ в течение 3 мин. теряется 80% активности факторов Villa и Va. Таким образом, тромбин по принципу положительной обратной связи не только ускоряет своё образование, но и, активируя протеин С, тормозит процесс свёртывания крови.
Наследственный дефицит протеина С и S ведёт к снижению скорости инактивации факторов VIlla и Va и сопровождается тромботической болезнью. Мутация гена фактора V, при которой синтезируется фактор V, резистентный к протеину С, также приводит к тромбогенезу.
Антикоагулянтная фаза вызывает торможение каскада реакций свёртывания крови, а ингибиторы ферментов свёртывания инактивируют активные ферменты в кровяном русле.
Ингибиторы ферментов свёртывания крови. Физиологические ингибиторы ферментов свёртывания крови ограничивают распространение тромба местом повреждения сосуда. Белок плазмы крови антитромбин III — наиболее сильный ингибитор свёртывания крови; на его долю приходится около 80 — 90% антикоагулянтной активности крови. Он инактивирует ряд сериновых протеаз крови: тромбин, факторы IХа, Ха, ХIIа, калликреин, плазмин и урокиназу. Антитромбин III не ингибирует фактор VIla и не влияет на факторы в составе мембранных комплексов, а устраняет ферменты, находящиеся в плазме крови, препятствуя распространению тромбообразования в кровотоке.
Взаимодействие антитромбина с ферментами свёртывания крови ускоряется в присутствии гепарина. Гепарин — гетерополисахарид, который синтезируется в тучных клетках. В результате взаимодействия с гепарином антитромбин III приобретает конформацию, при которой повышается его сродство к сериновым протеазам крови. После образования комплекса антитромбин III-гепарин-фермент гепарин освобождается из него и может присоединяться к другим молекулам антитромбина.
При наследственном дефиците антитромбина III в молодом возрасте наблюдают тромбозы и эмболии сосудов, опасные для жизни.
α2-Макроглобулин образует комплекс с сериновыми протеазами крови. В таком комплексе их активный центр полностью не блокируется, и они могут взаимодействовать с субстратами небольшого размера. Однако высокомолекулярные субстраты, например, фибриноген, становятся недоступными для действия протеаз в комплексе α2-макроглобулин-тромбин.
Антиконвертин (тканевый ингибитор внешнего пути свёртывания) синтезируется в эндотелии сосудов. Он специфически соединяется с ферментным комплексом Тф-VIIа-Са2+, после чего улавливается печенью и разрушается в ней.
α1-Антитрипсин ингибирует тромбин, фактор XIа, калликреин, однако он не рассматривается как важный ингибитор факторов свёртывания крови, α1-Антитрипсин в основном на тканевом уровне ингибирует панкреатические и лейкоцитарные протеазы, коллагеназу, ренин, урокиназу.
Пептиды, образующиеся в результате протеолитической активации проферментов и про- факторов, тоже обладают выраженными антикоагулянтными свойствами, но механизм их действия в настоящее время не выяснен.
Д. Роль тромбоцитов в гемостазе
Способность тромбоцитов прилипать к повреждённой поверхности стенки сосуда (адгезия) и друг к другу (агрегация), связываться с фибрином, образуя тромбоцитарный тромб, и секретировать в месте повреждения сосуда гемостатические факторы определяет их роль в гемостазе.
Циркулирующие в крови тромбоциты имеют дисковидную форму и не прилипают к неповреждённому эндотелию сосудов. Адгезию и агрегацию предотвращают взаимное отталкивание тромбоцитов и интактного эндотелия, а также простациклин (РG I2). Механизм действия некоторых индукторов и репрессора агрегации тромбоцитов рассмотрен на рис. 14-17.
Простациклин образуется из арахидоновой кислоты в эндотелиии сосудов и поступает в кровь (см. раздел 8). Синтез и секрецию простациклина эндотелиальными клетками стимулируют тромбин, гистамин, ангиотензин II и калликреин. Он реализует своё действие через аденилатциклазную систему передачи сигнала (см. раздел 5). Взаимодействие простациклина с рецептором вызывает активацию протеинкиназы А. Активная протеинкиназа А фосфорилирует и таким образом активирует
Са2+-АТФ-азу и Са2+-транслоказу. Это приводит к снижению уровня содержания Са2+ в цитоплазме тромбоцитов, сохранению ими дисковидной формы и снижению способности к агрегации.
Активация тромбоцитов сопровождается появлением на поверхности плазматической мембраны отрицательно заряженных участков, образованных фосфатидилсерином.
Основные индукторы активации и агрегации тромбоцитов — фактор фон Виллебранда, коллаген, тромбин, АДФ.
Фактор фон Виллебранда — гликопротеин, присутствующий в плазме крови, эндотелии сосудов и α-гранулах тромбоцитов. При повреждении стенки сосудов коллаген, базальная мембрана и миоциты субэндотелия взаимодействуют с тромбоцитами посредством фактора фон Виллебранда. Плазматическая мембрана тромбоцитов содержит несколько типов рецепторов этого фактора. Фактор фон Виллебранда, взаимодействуя с рецепторами, действует на тромбоциты через инозитолфосфатную систему передачи сигнала (см. раздел 5). В конечном итоге это приводит к повышению содержания Са2+ в цитоплазме тромбоцитов и образованию комплекса кальмодулин-4Са2+ — миозинкиназа. Фермент миозинкиназа в составе этого комплекса фосфорилирует сократительный белок миозин, который взаимодействует с актином с образованием актомиозина (тромбостенина). В результате этого тромбоциты приобретают шипо- видно-сферическую форму, облегчающую их взаимодействие друг с другом и с поверхностью повреждённого эндотелия.
Снижение концентрации фактора фон Виллебранда, уменьшение количества или изменение структуры его рецепторов ведут к нарушениям адгезии и агрегации тромбоцитов, что сопровождается кровоточивостью. Это наблюдают при синдроме Бернара—Сулье, обусловленном недостатком рецептора фактора фон Виллебранда гликопротеина Iа в тромбоцитах, и при болезни фон Виллебранда вследствие дефицита фактора фон Виллебранда.
Наиболее важные первичные индукторы активации тромбоцитов — тромбин и коллаген. Взаимодействие этих белков со специфическими рецепторами плазматической мембраны тромбоцитов приводит к мобилизации Са2+ из плотной тубулярной системы в цитоплазму, что в конечном итоге вызывает их адгезию и агрегацию.
Коллаген вызывает в тромбоцитах активацию фосфолипазы А2, которая освобождает арахидоновую кислоту из фосфолипидов их мембраны. Арахидоновая кислота служит субстратом для фермента циклооксигеназы (ЦОГ). В результате реакции, катализируемой циклооксигеназой, образуются циклические эндоперекиси простагландин С2(РG G2) и простагландин Н2(РG Н2). Эти простагландины под действием тромбоксансинтетазы превращаются в тромбоксан (см. раздел 8). Тромбоксан A2 снижает уровень цАМФ и, активируя фосфолипазу С, ускоряет освобождение Са2+ из плотной тубулярной системы (рис. 14-17).
Рис. 14-17. Механизм действия некоторых индукторов и репрессора агрегации тромбоцитов. Взаимодействие простациклина с рецептором R1, вызывает активацию аденилатциклазы, повышение концентрации цАМФ и вследствие этого снижение концентрации Са2+ в цитозоле тромбоцитов. Взаимодействие коллагена с рецептором Н4приводит к активации фосфолипазы А2, которая гидролизует фосфолипиды клеточных мембран с образованием арахидоновой кислоты. Арахидоновая кислота ферментом циклооксигеназой превращается в простагландины РGG2 и РGН2, из которых под действием тромбоксансинтетазы образуется тромбоксан А2. Тромбоксан А2 секретируется активированными тромбоцитами. Тромбоксан и тромбин, соединяясь с соответствующими рецепторами R3 и R2, активируют фосфолипазу С. Фосфолипаза С гидролизует мембранный фосфолипид фосфатидилинозитол 4,5-бисфосфат с образованием 1,4,5-инозитолтрифосфата и 1,2-диацилглицерола. Инозитолтрифосфат ускоряет поступление из плотной тубулярной системы в цитоплазму клетки Са2+, который соединяется с кальмодулином. В комплексе кальмодулин-4Са2+— миозинкиназа активная миозинкиназа фосфорилирует миозин. Фосфорилированный миозин взаимодействует с актином с образованием сократительного белка актомиозина, вызывающего изменение формы тромбоцитов, их адгезию и агрегацию. Комплекс протеинкиназа С-фосфатидилсерин-ДАГ-Са2+ фосфорилирует белок плекстрин. Фосфорилированный плекстрин вызывает освобождение содержимого плотных гранул и α-гранул тромбоцитов: АДФ, ГДФ, Са2+, серотонина, фактора фон Виллебранда, β-тромбомодулина. R1, R2, R3, — специфические мембранные рецепторы; АЦ — аденилатциклаза; G1, G2, G3, G4 — G-белки; ФЛС — фосфолипаза С; ФЛА2 — фосфолипаза А2; ФИФ2 — фосфатидилинозитол 4,5-бисфосфат; ИФ3 — 1,4,5-инозитол трифосфат; ДАГ — 1,2-диацилглицерол; ПКС — протеинкиназа С; ЦОГ — циклооксигеназа; ТхА2 — тромбоксан А2; ТхА2 синтетаза — тромбоксансинтетаза.
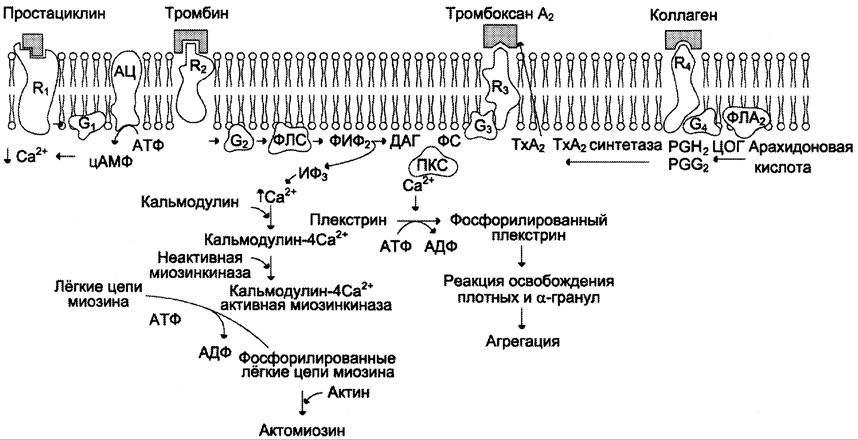
Тромбин взаимодействует со специфическим рецептором — интегральным белком, имеющим 7 трансмембранных доменов. Тромбин активирует рецептор частичным протеолизом, отщепляя от него N-концевой пептид, находящийся на внешней плазматической поверхности тромбоцита. Следовательно, тромбин, в отличие от других активаторов, действует каталитически, и одна молекула тромбина может активировать несколько рецепторов. Передача сигнала осуществляется через инозитолфосфатную систему, в результате чего в тромбоците повышается концентрация Са2+ и активируется протеинкиназа С. Образующийся комплекс кальмодулин — 4Са2+-миозинкиназа фосфорилирует миозин, взаимодействие которого с актином приводит к изменению формы тромбоцитов, к их адгезии и агрегации. Протеинкиназа С, кроме того, фосфорилирует белок тромбоцитов плекстрин. Фосфорилированный плекстрин вызывает «реакцию освобождения» содержащихся в гранулах тромбоцитов вторичных индукторов активации и агрегации тромбоцитов. К этим веществам относят содержащиеся в плотных гранулах тромбоцитов АДФ, Са2+, ГДФ, серотонин, гистамин и присутствующие в α-гранулах белок β-тромбоглобулин, фактор фон Виллебранда, белок фибронектин, тромбосподин и ВМК. Тромбосподин участвует во взаимодействии тромбоцитов друг с другом. β-Тромбоглобулин снижает секрецию простациклина и связывает гепарин. Фибронектин имеет центры связывания для коллагена, гепарина и тромбоцитов.
АДФ содержится в тромбоцитах, а также попадает в кровь при разрушении эритроцитов. АДФ взаимодействует со специфическими рецепторами и подавляет активность аденилатциклазы. Это вызывает увеличение мобилизации внутриклеточного Са2+ и в конечном итоге приводит к агрегации тромбоцитов.
Активация тромбоцитов, таким образом, сопровождается изменением их метаболизма и освобождением биологически активных веществ. Эти вещества вызывают морфологические изменения, адгезию, агрегацию тромбоцитов и участвуют в образовании тромба.
Нарушение функциональной активности рецепторов и системы вторичных посредников тромбоцитов приводит к изменению их функции и может явиться причиной ряда заболеваний, сопровождающихся тромбозами или кровотечениями.
Лекарственные препараты, нарушающие агрегацию тромбоцитов, используют для предупреждения возникновения тромбозов. Аспирин (ингибитор циклооксигеназы), никотиновая кислота (ингибитор тромбоксансинтетазы) и Са2+- блокаторы угнетают агрегацию тромбоцитов, влияя на разные этапы реализации тромбогенного сигнала.
Е. Фибринолиз
Тромб растворяется в течение нескольких дней после образования. Фибринолиз — ферментативное расщепление волокон фибрина с образованием растворимых пептидов, которые удаляются из сосудистого русла. Разрушение фибрина в составе тромба происходит под действием сериновой протеазы плазмина.
Плазмин образуется из плазминогена под действием активаторов. Неактивный профермент плазмина плазминоген синтезируется в печени, почках и костном мозге.
Тканевый активатор плазминогена (ТАП) — протеолитический фермент, содержащийся в эндотелии сосудов всех тканей, кроме печени. Поступление этого активатора в кровь увеличивается при эмоциональном напряжении, боли, венозной тромбоэмболии, умеренной физической работе. ТАП частичным протеолизом превращает неактивный плазминоген в активный плазмин. Активаторами плазминогена также служат фактор XIIа и калликреин.
Растворение фибринового сгустка происходит при взаимодействии фибрина, плазминогена и ТАП (рис. 14-18).
Рис. 14-18. Схема фибринолиза. 1 — абсорбированный на фибриновом сгустке плазминоген под действием активаторов (фактор ХIIа, калликреин, ТАП) частичным протеолизом превращается в плазмин; 2 — плазмин гидролизует фибрин с образованием растворимых пептидов X, Y, D, Е; 3 — в кровотоке ТАП инактивируется специфическими белками и-ТАП-1, и-ТАП-2; 4 — активность плазмина снижается под действием неспецифических ингибиторов сериновых протеаз (α2-антиплазмина, α2-макроглобулина, α1-антитрипсина, комплекса антитромбин-гепарин).
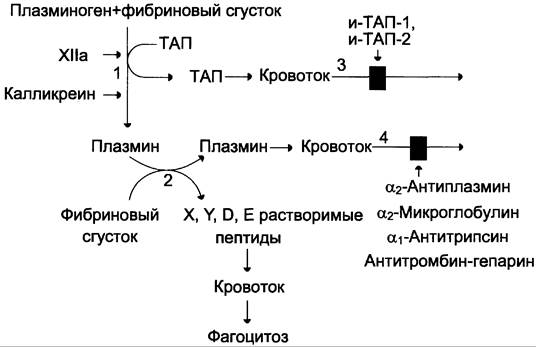
Формирование сети фибриновых волокон при образовании тромба сопровождается сорбцией на ней плазминогена и его активаторов. В молекуле плазмина и плазминогена есть участки, комплементарные доменам фибрина, причём одна молекула плазмина может связывать несколько молекул фибрина. Молекулы ТАП тоже имеют центры связывания с фибрином. Образующийся из плазминогена под действием ТАП плазмин гидролизует фибрин с образованием пептидов X и Y, активирующих фибринолиз, и пептидов D и Е, его тормозящих. Растворимые пептиды X, Y, D, Е поступают в кровоток и там фагоцитируются. Разрушение тромба приводит к освобождению из него плазмина и ТАП. В кровяном русле последние быстро инактивируются специфическими ингибиторами и улавливаются печенью.
ТАП ингибируется ингибиторами тканевого активатора плазмина первого (и-ТАП-1) и второго (и-ТАП-2) типов, а плазмин — α2-антиплазмином или другими ингибиторами сериновых протеаз.
В почках синтезируется протеолитический активатор плазминогена урокиназа, которая, превращая плазминоген в плазмин, способствует освобождению почечных клубочков от фибриновых волокон.
Из β-гемолитического стрептококка выделили белок стрептокиназу, образующий комплекс с плазминогеном, в котором плазминоген аутокаталитически превращается в плазмин.
Урокиназу, стрептокиназу и ТАП используют при тромболитической терапии инфаркта миокарда, тромбозах вен и артерий, гемодиализе.
Такие ингибиторы ферментов свёртывания крови, как α2-макроглобулин, α1-антитрипсин и комплекс антитромбин III-гепарин также обладают небольшой фибринолитической активностью.
Снижение фибринолитической активности крови сопровождается тромбозами. Нарушение разрушения фибринового сгустка может быть вызвано наследственным дефицитом плазминогена или генетическим дефектом его структуры, снижением поступления в кровь активаторов плазминогена, повышением содержания в крови ингибиторов фибринолиза (и-ТАП-1, и- ТАП-2, α2-антиплазмина).
Наследственные и приобретённые нарушения гемостаза могут привести как к геморрагическим заболеваниям, характеризующимся кровоточивостью, так и к тромботической болезни. Однако следует отметить, что повышенная склонность к тромбообразованию и внутрисосудистому свёртыванию (тромбофилии) встречается гораздо чаще, чем гемофилии. Например, частота разных форм гемофилий колеблется в разных странах от 6 до 18 на 100 000 мужчин, в то время как тромбофилии, вызванные дефицитом антитромбина III, встречаются у 1-2 больных на 5000, а при недостатке протеина С — у одного на 15 000 человек.