ЕКОЛОГІЧНА БІОХІМІЯ - Навчальний посібник - В. М. Ісаєнко 2005
Розділ 10. БІОТРАНСФОРМАЦІЯ КСЕНОБІОТИКІВ
10.1.Окиснення, відновлення, деградація і кон'югація ксенобіотиків
Реакції, які беруть участь у біотрансформації ксенобіотиків, у тому числі забруднювачів довкілля, можна умовно розподілити на чотири класи: 1) окиснення; 2) відновлення; 3) деградації; 4) кон’югації.
Реакції окиснення в мікросомах. Серед великої кількості реакцій окиснення ксенобіотиків основними є: 1) окиснення спиртів і альдегідів; 2) окиснення амінів, гідразинів, азиридинів; 3) окиснення ароматичних алкілзамінних сполук; 4) гідроксилювання кільцевих структур; 5) ароматизація аліциклічних сполук; 6) епоксидація; 7) окиснення або окисне заміщення органічної сірки; 8) окисне дезалкилювання.
Зазначимо, що за окисної біотрансформації ксенобіотиків можуть утворюватися токсичні, мутагенні або канцерогенні сполуки.
Серед клітинних систем, які беруть участь в окисненні ксенобіотиків, особливе місце посідають мікросоми. Ферменти мікросом, що використовують молекулярний оксиген для окиснення ксенобіотиків, поділяють на монооксигенази, диоксигенази та оксидази.
Монооксигенази приєднують один атом оксигену від донора гідрогену (ДНг) до субстрату (S) і відновлюють другий атом до води (рис. 10.1).
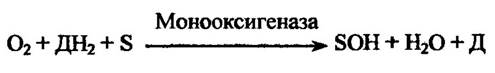
Діоксигенази приєднують до субстрату два атоми оксигену:
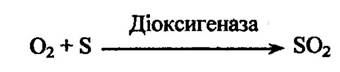
Оксидази відновлюють молекулу оксигену до пероксиду гідрогену або двох молекул води без упровадження оксигену в субстрат:
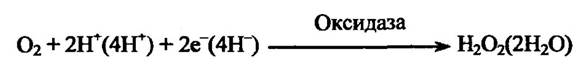
Монооксигенази найчастіше каталізують реакції пдроксилювання ксенобіотиків. Вони поділяються на дві групи — ті, які не містять металів, і ті, що містять один або кілька атомів металу, — метало-ферменти. В останніх, як правило, містяться атоми купруму (тирозиназа, допамін-β-монооксигеназа та ін.) або феруму (фенілаланін- гідролаза тощо). До цієї трупи належать також монооксигенази, які містять як простетичну групу цитохром Р450. У свою чергу, їх можна розподілити на три підгрупи залежно від місця локалізації (рис. 10.1).

Рис. 10.1. Локалізація та способи відновлення О2 монооксигеназами, які містять цитохром P450
У ссавців центральне місце в процесі біотрансформації ксенобіотиків належить мікросомам гепатоцитів, які становлять до 25 % сухої маси клітин.
Монооксигенази мікросом печінки ссавців містять принаймні НАДФН— цитохром Р450— оксидредуктазу, НАДН— цитохром b5 — оксидоредуктазу і цитохром Р450.
Каталітична активність НАДФН— цитохром Р450— оксидоредуктази залежить від способу виділення з мікросом. Так, якщо цю ферментну систему солюбілізувати детергентами, то вона безпосередньо здатна відновлювати цитохром с, а за наявності ліпідів — цитохром Р450. Солюбілізована трипсином, ця оксидредуктаза відновлює цитохром с, але не цитохром Р450, навіть коли до ферменту додають фосфоліпази. Оскільки фермент значною мірою споріднений із цитохромом с, то його часто називають також НАДФН — цитохром с — оксидоредуктазою. Проте відомо, що цитохром с локалізований в основному в мітохондріях і його дуже мало в мікротомах і цитозолі. Тому цитохром с є звичайним акцептором електронів для даної оксидоредуктази.
Основне призначення НАДФН — цитохром Р450 — оксидоредуктази в гідроксилюючому комплексі мікросом клітин — передавання електронів на цитохром Р450 і, отже, окиснення ксенобіотиків. Цей фермент може брати участь також у нітрогенредуктазній реакції, окисненні жирних кислот, N-окисненні амінів тощо.
Мікротоми також містять НАДН-залежну електрон-транспортну систему, початковою ділянкою якої є НАДН — цитохром b5 — оксидо-редуктаза (разом з ФАД), розміщена в мембрані поряд із цитохром Р450.
Показано, що цитохром 65 здатний за певних умов відновлювати не тільки НАДН, а й НАДФН. Перенесення електронів з НАДН-залежного ланцюга в ланцюг окиснення НАДФН може відбуватися трьома способами:
1) безпосередньо з НАДН-специфічного ферменту на цитохром Р450
2) з цитохрому b5 ланцюга окиснення НАДН на цитохром b5 ланцюга окиснення НАДФН;
3) з цитохрому Ь5 ланцюга окиснення НАДН на цитохром Р450.
Цитохром Р450 гемопротеїном, у якого гемін (хелаторний комплекс Fe3+—Р450(Fе3+)) у результаті приєднання електрона від донора перетворюється в протогем (хелаторний комплекс Fe2+— Р450(Fе2+)). Крім печінки, він локалізований також у мікротомах легень, нирок, селезінки, слизовій оболонці кишковику, а також плаценті та деяких інших органах. Цей цитохром знайдено у ссавців, деяких видів птахів, земноводних, безхребетних, бактерій і вищих рослин.
Цитохром Р450 в гідроксилюючому комплексі взаємодіє із субстратом і молекулярним оксигеном, приймає електрони від відповідних донорів.
На першому етапі відбувається взаємодія цитохрому Р450(Fe3+) із субстратом (S) — ксенобіотиком з утворенням комплексу цитохром
Р450(Fе3+) — субстрат. На другому етапі комплекс відновлюється в НАДФН-специфічному ланцюгу перенесення електронів з утворенням комплексу Р450(Fе2+) — субстрат. На наступному етапі відбувається взаємодія атмосферного оксигену з комплексом Р450(Fе3+) — субстрат з утворенням комплексу Р450(Fе3+) — субстрат — О2. У подальшому відбувається активування молекулярного оксигену в оксигенованому комплексом його відновленням. Оксигенований комплекс відновлюється в реакції одноелектронного перенесення (P450(Fe2+) → Р450(Fе3+)). Другий електрон потрапляє з ланцюга окиснення НАДН, і його перенесення відбувається за участю цитохром b5 (НАДН — цитохром b5 — оксидоредуктази). На четвертому етапі відбувається розпад утвореного комплексу на окиснений цитохром Р450 і окиснений субстрат, при цьому один атом оксигену переноситься на субстрат, а інший відновлюється до води. На останньому, п’ятому, етапі нестійкий комплекс P450(Fe3+) — окиснений субстрат дисоціює з вивільненням цитохрому Р450(Fе3+), який знову вступає в повторну реакцію.
Один із можливих способів гідроксилювання ксенобіотиків за участю цитохрому Р450 наведено на рис. 10.2.
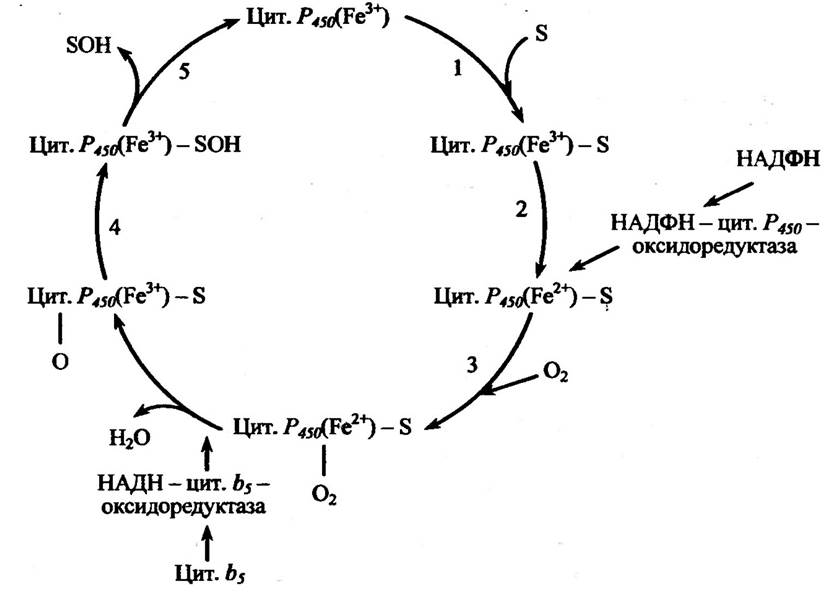
Рис. 10.2. Схема реакції гідроксилювання ксенобіотиків за участю цитохрому Р450:
цит. Р450—цитохром Р450; цит. b5 — цитохром b5
Ксенобіотики, які здатні індукувати гідроксилюючий комплекс з цитохромом Р430, можна розподілити на три групи: 1) фенобарбітал, дифенілгідантоїн, пестициди та деякі інші, що збільшують активність НАДФН — цитохром Р450— оксидоредуктази та самого цитохрому P450, 2) поліциклічні вуглеводи та деякі інші речовини не впливають на активність НАДФН— цитохром Р450— оксидоредуктази і на швидкість відновлення цитохрому Р45, 3) стероїди, їхні синтетичні аналоги та подібні до них речовини, що підсилюють активність НАДФН— цитохром Р4 Р450— оксидоредуктази та збільшують швидкість відновлення цитохрому Р450 без зміни кількості останнього.
Деякі ксенобіотики здатні інгібувати метаболізм інших чужорідних сполук і, як наслідок, пролонгувати їхню дію. До таких сполук належать β-діетиламіноетилдифенілпропілацетатгідрохлорид (SKF525A), 2,4-дихлор-6-фенілфеноксиетиламін (ДФЕФ), N-метил-З-пипередил-(N',N) дифеніл карбомат (МПДК), іпроніазид, хлорциклізин, глутетемід і т. ін. Інгібітори, як правило, безпосередньо діють на ферменти окиснення ксенобіотиків на відміну від індукторів, які впливають на їх синтез.
Немікросомальне окиснення. Крім мікросомальних оксигеназ у мітохондріях, розчинних клітинних фракціях та плазмі крові виявлено ферментні системи, які здатні каталізувати реакції окиснення ксенобіотиків, основними з яких є окисне дезамінування, окиснення спиртів та альдегідів, ароматизація аліциклічних сполук. Деякі з цих ферментів можуть каталізувати також зворотні перетворення, тобто відновлення продуктів окиснення ксенобіотиків.
У процесі окисного дезамінування аліфатичні аміни оксигенуються амінооксидазами у відповідні альдегіди відщепленням аміаку (рис. 10.3).
![]()
Рис. 10.3. Реакція окиснення аліфатичних амінів у процесі окисного дезамінування
Деякі з цих ферментів мають відмінності щодо субстратної специфічності, дії інгібіторів і локалізації в клітинах. Вони локалізовані в печінці, нирках, слизовій оболонці кишечнику та плазмі крові.
Моноаміноксидаза, локалізована в мітохондріях, каталізує окисне дезамінування первинних, вторинних і третинних аліфатичних амінів. Первинні аміни метаболізуються у відповідну кислоту або спирт через альдегід. Вторинні та третинні аміни стійкіші до дезамінування й переважно підлягають дезалкілюванню, утворюючи первинні аміни, або виділяються незмінними. Крім екзогенних амінів, моноаміноксидаза дезамінує також природні аміни, такі як 5-окситриптамін, катехоламіни та їхні метальні похідні, а також деякі інші сполуки.
Діаміноксидазу виявлено в багатьох клітинах, зокрема в печінці, нирках, слизовій оболонці кишечнику, однак вона локалізується переважно в мітохондріях. Діаміноксидаза окисно дезамінує діаміни (гістидин, кадаверин, путресцин та ін.) шляхом видалення однієї молекули аміаку.
Ця ферментна система не дезамінує діаміни з дев’ятьма або більше атомами карбону. Замість цього вони дезамінуються моноамі- ноксидазою, яка, у свою чергу, не дезамінує нижчі діаміни.
У плазмі крові виявлено кілька амінооксидаз. Так, наприклад, спермінооксидаза окисно дезамінує спермін та інші поліаміни, а бен- зиламінооксидаза — бензиламін і мескалін.
Алкогольдегідрогеназа, локалізована в розчинній фракції печінки, нирок і легенів, оксигенує первинні спирти, такі як етанол, н-бутанол, фторетанол, бензиловий спирт, циклогексаль. Так, у випадку етанолу відбувається така реакція (рис. 10.4).

Рис. 10.4. Реакція дегідрування етанолу алкогольдегідрогеназою
Варто зазначити, що окиснення етанолу може виконувати також специфічна мікросомальна етанолооксигенуюча система (використовує НАДФ Н), а також (певною мірою) каталаза (використовує гідропероксид).
Може відбуватися і зворотна реакція, в якій альдегіди й кетони (ацетальдегід, ацетон, циклогексан та ін.) відновлюються в спирти.
Алкогольдегідрогеназа ссавців має низьку спорідненість із метанолом, який метаболізується в основному пероксидазами, зокрема ксантиноксидазою й каталазою.
Вторинні спирти в організмі тварин оксигенуються в кетони алкогольдегідрогеназою, але швидкість їх окиснення значно менша, ніж первинних. Вищі вторинні спирти та третинні спирти окиснюються надзвичайно повільно.
Аліфатичні й ароматичні альдегіди оксигенуються у відповідні карбонові кислоти, які в подальшому знову можуть піддаватися перетворенню в процесі β-окиснення. До ферментів, які каталізують такі реакції у ссавців, належать альдегідоксидаза, ксантиноксидаза і НАД-специфічна альдегіддегідрогеназа.
Альдегідоксидаза та ксантиноксидаза локалізовані в розчинній фракції клітин печінки. Ці ферменти каталізують окиснення альдегідів, які утворюються під час дезамінування ендогенних амінів (адреналіну, норадреналіну, 5-окситриптаміну) та чужорідних амінів (бензальдегіду, ацетальдегіду та саліцилальдегіду). На рис. 10.5 наведено реакцію окиснення бензальдегіду.
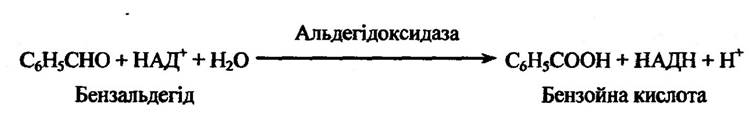
Рис. 10.5. Окиснення бензальдегіду альдегідоксидазою
Залежна від НАД+ альдегіддегідрогеназа каталізує окиснення хлоральгідраіу в трихлороцтову кислоту (рис. 10.6).

Рис. 10.6. Реакція дегідрування хлоральгідрату альдегідцегідрогеназою
У мітохондріях міститься ферментна система, яка каталізує перетворення циклогексанкарбонові кислоти в ароматичні кислоти. У результаті цього може утворюватися бензойна кислота (рис. 10.7).
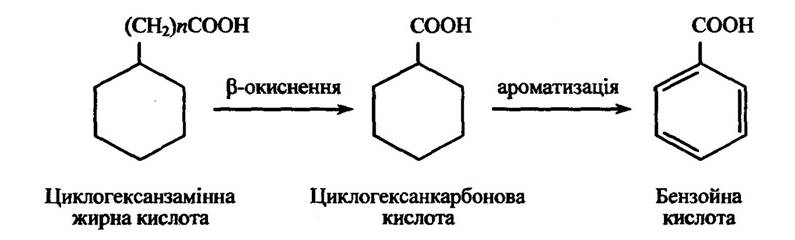
Рис. 10.7. Реакція окиснення циклогексанзамінних жирних кислот з парним значенням = 2,4,6 ...)
Якщо циклогексанзамінні жирні кислоти (загальна формула С6Н11(СН2)nСООН) містять непарне число n, то вони перетворюються в циклогексаноцтову кислоту, яка потім метаболізується завдяки окисненню циклогексанового кільця.
Ферментну систему, яка каталізує ароматизацію ациклічних сполук, виявлено в мітохондріях печінки та нирок, вона найактивніша у кролів, мурчаків і зовсім неактивна в мишей, собак і людини.
Реакції відновлення в мікросомах. Ці реакції за біотрансформації ксенобіотиків поширені менше, ніж окиснення. Водночас їхній внесок у загальну біотрансформацію ксенобіотиків істотний.
Нітратредуктази мікросом каталізують відновлення ароматичних нітросполук їхня ферментативна активність залежить від багатьох факторів, і насамперед від наявності в середовищі НАДН і НАДФН, малоспецифічна до субстрату та відновлює майже всі ароматичні аміни. У реакції відновлення нітросполук беруть участь флавопротеїн і цитохром Р450.
Початкова стадія відновлення субстратів пов’язана, мабуть, із транспортом відновлених еквівалентів від флавопротеїну, наслідком якою є утворення проміжних нітропохідних сполук і гідроксиламі- ну (рис. 10.8).
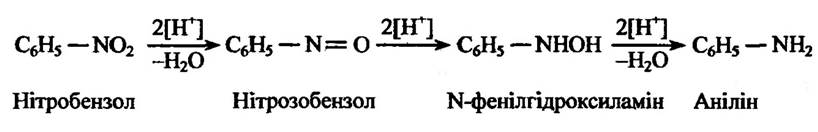
Рис. 10.8. Реакція відновлення ароматичних нітросполук нітратредуктазою
Разом з тим, очевидно, що цитохром Р450 передає електрони проміжним сполукам, які утворюються після виникнення нітробензоль- них аніонних радикалів. Відновлення амінів нітроредуктазою відбувається у дві стадії—відновлення субстратів до гідрозосполук та їх відновне розщеплення (рис. 10.9).
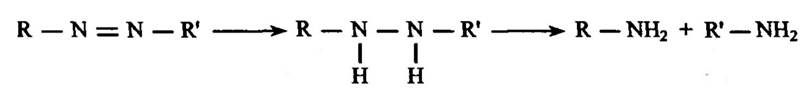
Рис. 10.9. Реакція відновлення нітрозосполук азотредуктазою
Нітросполуки в мікросомах можуть відновлюватися за участю НАДФН — цитохром Р450 — оксидоредуктази, брати участь також можуть НАДФН — цитохром b5 — оксидоредуктаза, цитохром Р450 і флавопротеїн. Одне з припущень — НАДФН — цитохром Р450 — оксидоредуктаза передає електрони на субстрат за участю флавонів, які цілком або наполовину відновлюють цитохром с.
У мікросомах печінки локалізовані N-оксидоредуктази, які відновлюють N-окиси до відповідних амінів. Вони проявляють активність і за відсутності НАДФН— цитохром — оксидоредуктази та цитохрому Р450.
Досліджено також відновлення нітрозосполук у мікросомах ніт- роредуктазами. Активність цих ферментів найвища в печінці та значно менша в нирках, легенях, слизовій оболонці тонкого кишечнику.
Епоксидредуктаза каталізує процес відновлення ароматичних епоксидів. Для проявлення її активності необхідний НАДФН, і цей процес забезпечується цитохром Р450 -залежними ферментами.
У мікросомах печінки міститься також фермент, який за наявності НАДФН і оксигену видаляє галоген з його аліфатичної частини з одночасним відновленням субстрату. Так, наприклад, хлороформ відновлюється так (рис. 10.10).
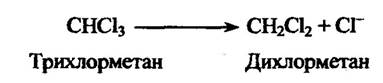
Рис. 10.10. Реакція відновлення хлороформу
Можливе також видалення флуору. Типовим прикладом такого процесу є дегалогенування галатону, результатом якого є елімінування атома флуору (рис. 10.11).
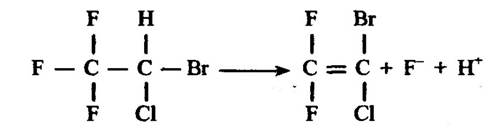
Рис. 10.11. Реакція відновлення галатону
Відновлення кетонів до спиртів каталізується кетонредуктазами. Ця реакція, як привило, є оборотною.
Немікросомальне відновлення. Ряд ферментів, які беруть участь у реакціях відновлення ксенобіотиків, локалізовані також поза межами мікросом. Так, у цитоплазмі містяться нітроредуктази і кетоноредуктази, у мітохондріях і плазмі крові— N-оксидоредуктази тощо. Багато ферментів, які каталізують реакції відновлення, містяться в мікроорганізмах.
Відновлення подвійних зв’язків спостерігаються в метаболізмі ряду ненасичених аліфатичних або аліциклічних сполук, які здатні ставати насиченими. До таких сполук належать моноциклічні терпени. Прикладом відновлення подвійного зв’язку циклогександієнового кільця є перетворення αа-феландрену (ментадієну) у феландре- нову кислоту (параментан).
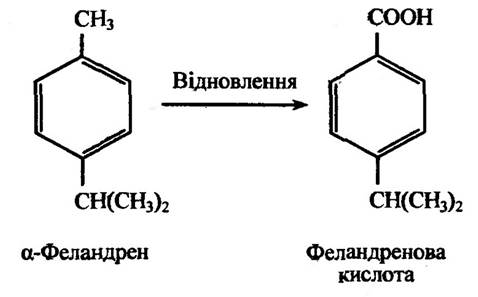
Рис. 10.12. Реакція відновлення β-феландрену
Відновлення подвійного зв’язку в бічному ланцюзі відбувається за перетворення пулеголу в ментол (рис. 10.13).
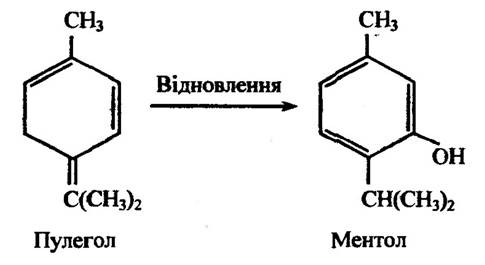
Рис. 10.13. Реакція відновлення пулеголу
Одним із типів немікросомального відновлення є відновлення дисульфідів у тіоли. Діетилсульфіди при цьому відновлюються в етилмеркаптан (рис. 10,14).
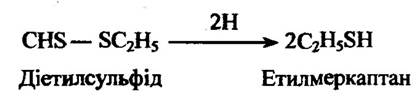
Рис. 10.14. Реакція відновлення діетилсульфіду
Гідроксамові кислоти здатні до відновлення у відповідний амід. Так, наприклад, саліцилгідроксамова кислота перетворюється в салі циламід (рис. 10.15).
У свою чергу, саліциламід утворює кон’югати з 5-бромсаліцил-амідом, який утворюється під час відновлення 5-бромпохідної саліцилгідроксамової кислоти.
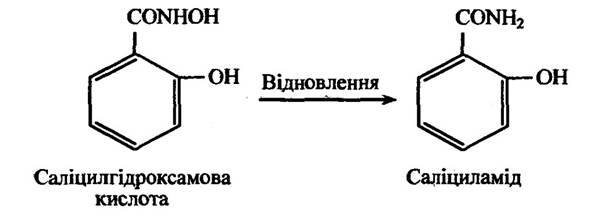
Рис. 10.15. Реакція відновлення саліцилгідроксамової кислоти
Сульфоксиди та N-оксиди теж здатні до відновлення. Так, наприклад, диметилсульфоксид відновлюється в диметилсульфід, a N- окситриметиламіну —у триметиламін (рис. 10.16).
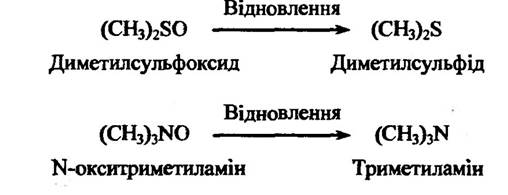
Рис. 10.16. Реакція відновлення диметилсульфоксиду та N-окситриметиламіну
Ще один із типів немікросомального відновлення — це відновне дегідроксилювання гідроксамових кислот, катехолів і деяких аліфатичних гідроксильних похідних.
У процесі ароматичного дегідроксилювання одним зі способів є видалення 4-гідроксильної групи фенольного кільця. Так, наприклад, ароматичне дегідроксилювання гомопротокатехової кислоти (З, 4-діоксифенілоцтової кислоти) відбувається з утворенням мета- або параоксифеліоцтових кислот (рис. 10.17).
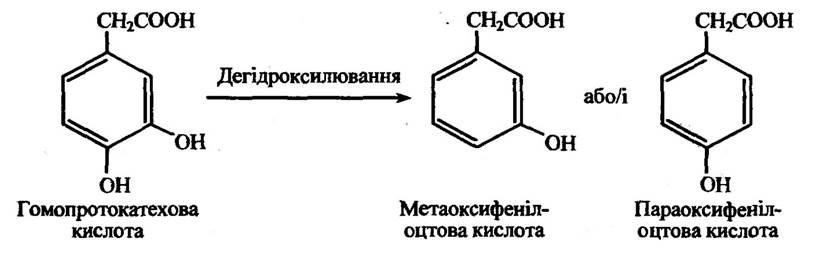
Рис. 10.17. Реакція дегідроксилювання гомопотокатехової кислоти
Так само може дегідроксилюватися 3,4-діоксикорична кислота (кавова кислота), 3,4-діоксифеніланін (ДОФА), флавоноїди рутину й кварцетину та інші похідні пірокатехіну.
Установлено, що крім дегідроксилювання 4-заміщених похідних пірокатехіну, втрата фенольної гідроксильної групи може відбуватися і в ксантуренової кислоти, яка здатна перетворюватися в 8-оксихінальдинову (рис. 10.18).
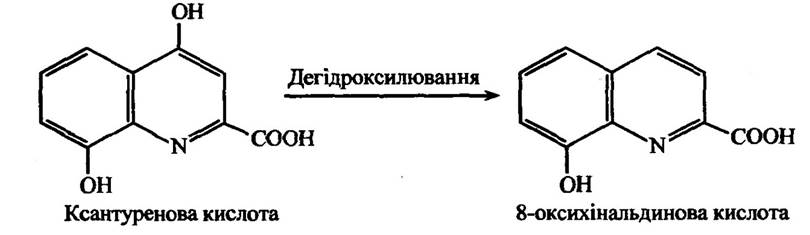
Рис. 10.18. Реакція дегідроксилювання ксантуренової кислоти
Аліфатичне дегідроксилювання норадреналіну, як і ароматичне, приводить до утворення поряд з іншими метаболітами метаоксифенілоцтової та гомованілінової кислот (рис. 10.19).
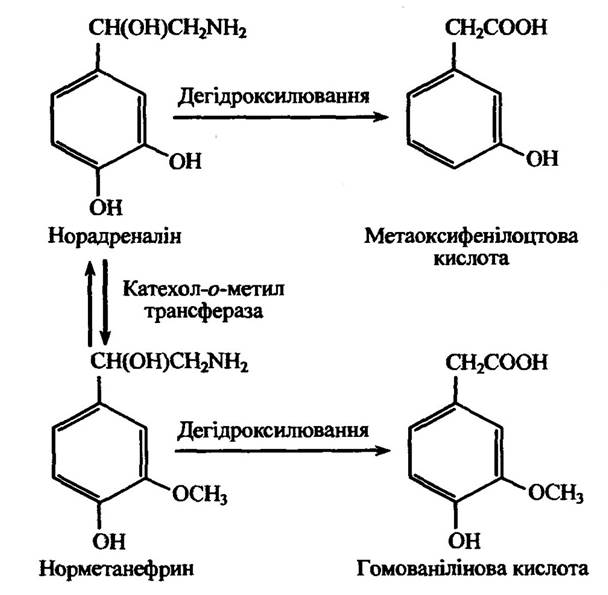
Рис. 10.19. Реакція дегідроксилювання норадреналіну
Одним із типів дегідроксилювання є також N-дегідроксилювання. Типовим прикладом такого процесу є перетворення N-оксиуретану в уретан (рис. 10.20).
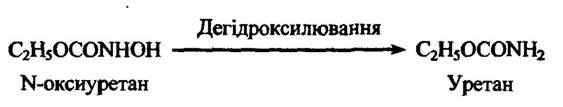
Рис. 10.20. Реакція дегідроксилювання N-оксиуретану
Подібний процес може відбуватися й під час перетворення 4-гідроксил-амінобіфенілу (перетворюється в 4-амінобіфеніл), N-окси-4-ацетиламіностильбену (перетворюється в 4-ацетиламіностильбен), N-окси-2-N-ацетиламінофлюорену (перетворюється в 2-ацетиламінофлюорен) та інших сполук.
Внесок у реакцію відновлення за біотрансформації ксенобіотиків роблять також процеси відновлення атомів зі змінною валентністю.
Реакції дегідратації. Прикладами реакцій дегідратації (розщеплення) ксенобіотиків є гідроліз етерів. Ці реакції каталізуються поширеними ферментами — холінестеразами, аліестеразами, арилестиразами тощо. Гідролізу підлягають деякі алкалоїди— атропін, кокаїн та ін. Зокрема, кокаїн гідролізується ферментами плазми кролів, а не людини (рис. 10.21).
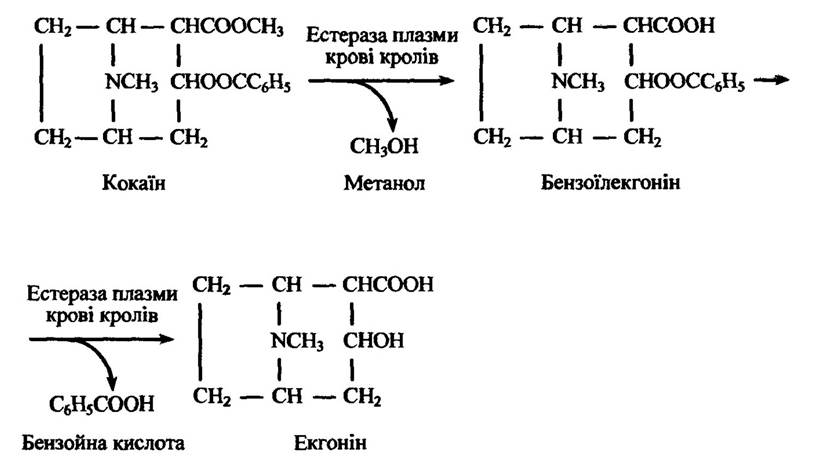
Рис. 10.21. Реакція гідролізу кокаїну
Естерази мікросом печінки здатні гідролізувати ряд етерів, зокрема меперидин (рис. 10.22).
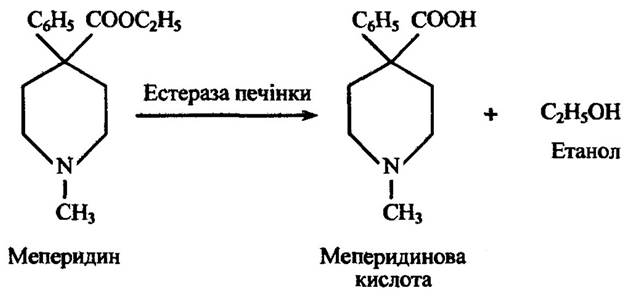
Рис. 10.22. Реакція гідролізу меперидину
Аміди в організмі тварин стійкіші, ніж відповідні складні етери. Тому їх гідроліз відбувається повільніше.
Швидкість гідролізу аліфатичних амідів залежить від довжини ланцюга алкільної групи. Так, наприклад, фенілацетатамід гідролізується відносно швидко, а ацетанілід—лише в невеликій кількості і в основному виводиться з організму незмінним.
Гідроліз ароматичних амідів теж залежить від їхньої хімічної природи та положення замісників в ароматичному кільці. Так, наприклад, бензамід (C6H5CONH2) гідролізується цілком, а параамінобензамід—лише на 20 %, параоксибензамід — на 4 %.
Гідроксильовані кислоти можуть метаболізуватися не тільки відновленням, як це вже було розглянуто, а й гідролізуватися в ароматичну карбонову кислоту. Так, наприклад, бензгідроксамова кислота здатна гідролізуватися до бензойної кислоти (рис. 10.23).
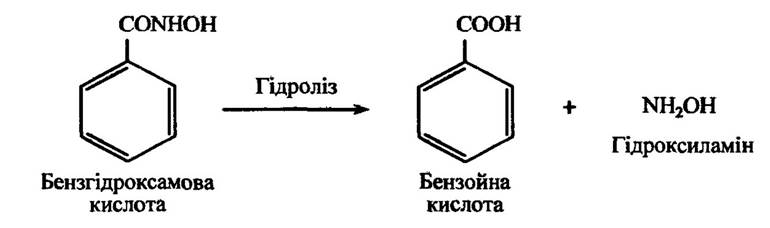
Рис. 10.23. Реакція гідролізу бензгідроксамової кислоти
Гідразиди ароматичних кислот гідролізуються аналогічно. Прикладом такого процесу може бути гідроліз бензгідрозиду в бензойну кислоту (рис. 10.24).
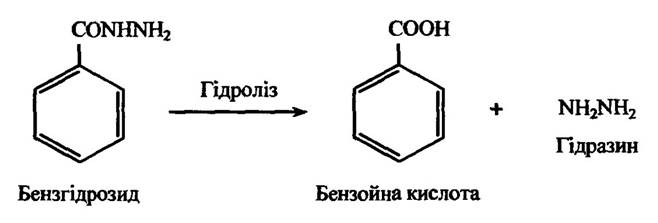
Рис. 10.24. Реакція гідролізу бензгідрозиду
Карбомати теж здатні гідролізуватися з утворенням карбомінової кислоти та спирту (рис. 10.25).
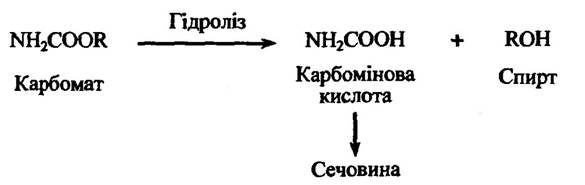
Рис. 10.25. Реакція гідролізу карбомату
Так гідролізуються також етилкарбомати, зокрема наркотик гедонал (метилпропінілкарбінілкарбомат).
Замінні уретани також здатні гідролізуватися, зокрема етилхлорамат — до трихлороцтової кислоти.
Нітрили або ароматичні ціаніди метаболізуються в основному шляхом ароматичного гідроксилювання. Разом з тим вони можуть гідролізуватися з утворенням відповідної карбонової кислоти. Так, наприклад, гідроліз бензонітрилу призводить до утворення бензойної кислоти (рис. 10.26).
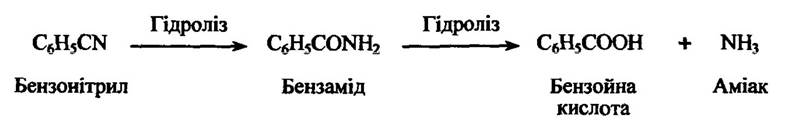
Рис. 10.26. Реакція гідролізу бензонітрилу
Ряд гетероциклічних сполук гідролізуються шляхом розриву гетероциклічного кільця. Найпростішими такими сполуками, очевидно, є гідантоїни. Реакцію гідролізу гідантоїну ілюструє рис. 10.27.

Рис. 10.27. Реакція гідролітичного розщеплення гідантоїну
Бензоксазоли здатні гідролізуватися шляхом розриву оксазольного кільця з утворенням ортоформамідофенольних похідних з подальшим утворенням ортоамінофенольних похідних (рис. 10.28).
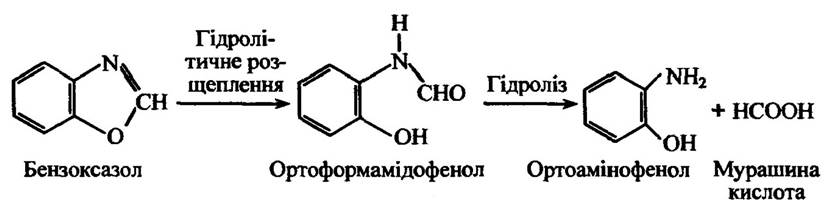
Рис. 10.28. Гідролітичне розщеплення бензоксазолу
Індол спочатку гідроксилюється з утворенням індоксилу, а відтак — ізатеніну, в якому відбувається гідролітичне розщеплення пі- рольного кільця з утворенням в остаточному підсумку антранілової та мурашина кислоти (рис. 10.29).
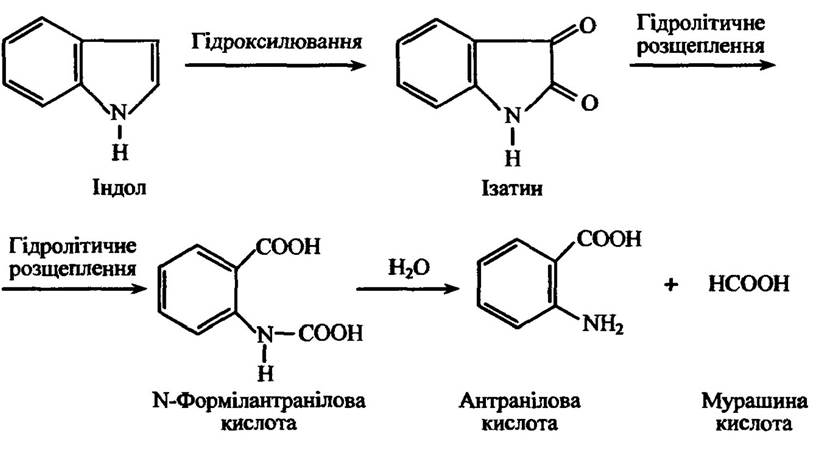
Рис. 10.29. Реакції метаболізу ендолу
Подібним чином відбувається розкриття гетероциклічного кільця в процесі метаболізму кумарину. Відбувається гідроксилювання кумарину з утворенням 3-оксикумарину та його гідролітичне розщеплення до ортооксифенілоцтової та (або) ортооксилмолочної кислоти (рис. 10.30).
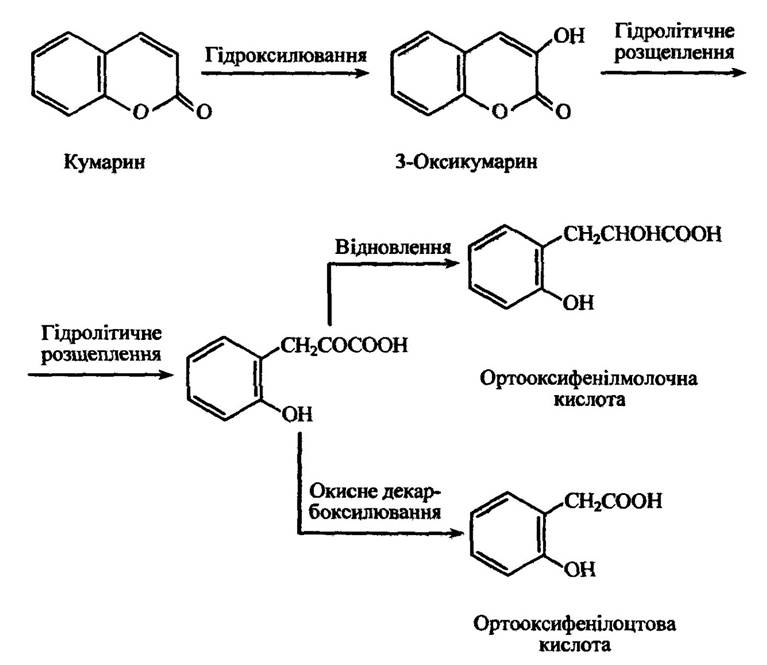
Рис. 10.30. Реакції метаболізму кумарину
У фловоноїдів також може відбуватися розкриття гетероциклічного кільця, місце якого визначається видом тварин і замісниками в пірановому кільці. Реакції гідролітичного розщеплення діосметину та кварцетину наведено на рис. 10.31.
В ароматичних сполук може відбуватися також розкриття кілець шляхами, які відмінні від уже розглянутих. Таким прикладом може бути метаболізм бензолу, який приводить до утворення муконової кислоти (рис. 10.32).
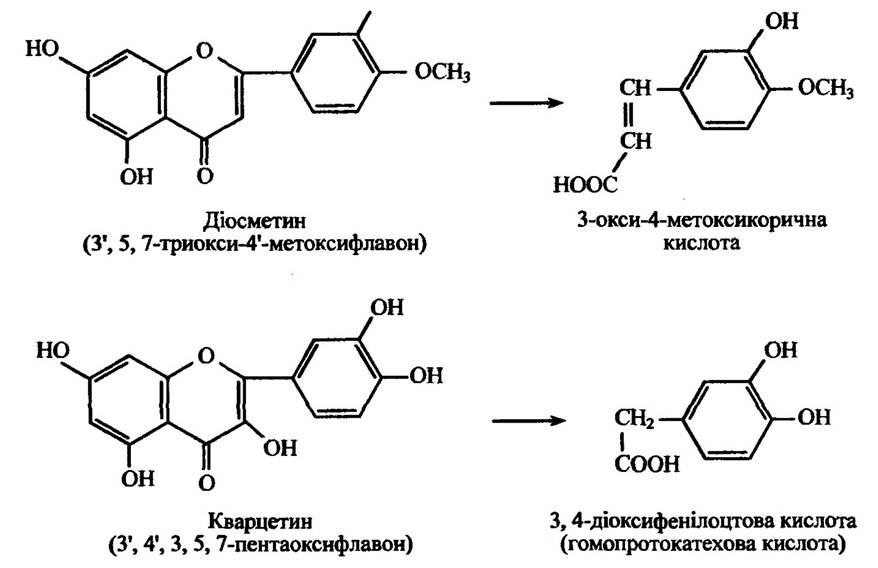
Рис. 10.31. Реакції метаболізму діосметину та кварцетину
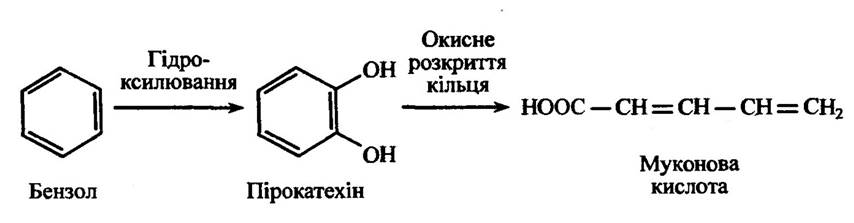
Рис. 10.32. Метаболізм бензолу з утворенням муконової кислоти
Існує ціла низка способів розщеплення циклічних структур. Істотне значення має біодеградація лигніну— природного полімеру, вміст якого в деревині становить 20—30 % сухої маси.
На першому етапі деградації лигніну мікроорганізмами відбувається деметилювання метоксильних груп гвайацильних і сирингіль- них одиниць полімеру лигніну. Наслідком цього є зменшення вмісту метоксильних груп і збільшення орто- та дифенольних залишків. На наступному етапі в цих залишках під дією діоксигеназ грибів відбувається розкриття ароматичного кільця лигніну з утворенням аліфатичних карбоксильних груп.
Арсенобензолові похідні здатні метаболізуватися розривом молекул в окисній реакції, утворюючи спочатку арсеноксиди, а відтак арсенокислоти. Прикладом таких реакцій може бути розщеплення протисифілічного препарату арсфенаміну (рис. 10.33).
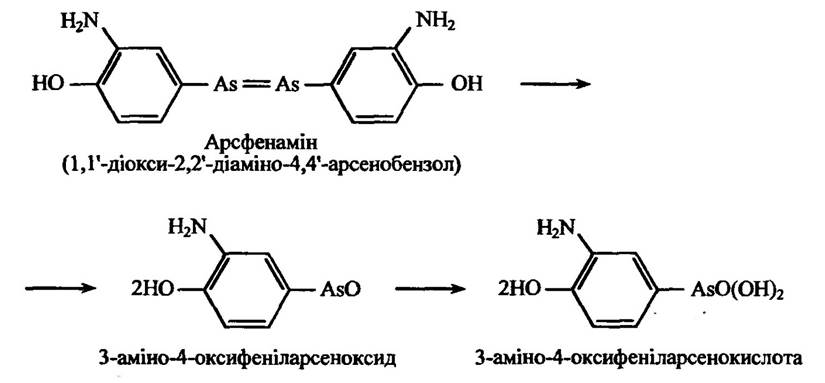
Рис. 10.33. Реакція окисного розщеплення арсфенаміну
На відміну від арсенобензолових похідних азобензолові похідні розщеплюються не під час окиснення, а в процесі відновлення.
Один зі способів деградації сполук — циклізація. Прикладом таких процесів може бути перетворення ортоамінофенілетанолу та різних ортонітрофенілових похідних (ортонітрофенілацетилену, ортонітрофенілпропіонової кислоти та ін.) (рис. 10.34).
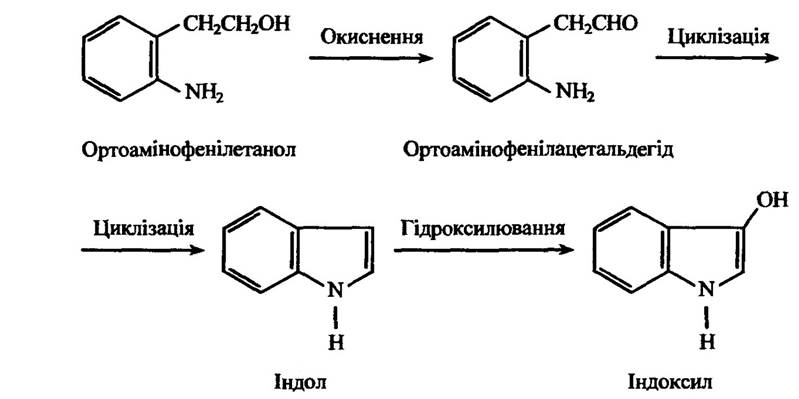
Рис. 10.34. Реакція циклізації ортоамінофенілетанолу
Ортокумаринова (ортоокситранскорична) та ортогідрокумаринова (ортооксифенілпропіонова) кислоти циклізуються аналогічно.
Певні мііфоорганізми здатні дегалогенувати хлорвмісні ксенобіотики, до яких належать багато речовин, у тому числі пестициди та ряд природних метаболітів нижчих рослин.
Найістотнішими є окисне, відновне та гідролітичне дегалогенування. Окисне дегалогенування відбувається за механізмами дегідрогалогенування, окисного дегалогенування з утворенням подвійних зв’язків та дегалогенування — гідроксилювання за участю молекул оксигену монооксигеназами та діоксигеназами.
Монооксигенази здатні брати участь у деградації мікроорганізмами хлорованих феноксіалканових кислот, можуть відщеплювати від ароматичних кілець флуор у процесі гідроксилювання тощо. Діоксигенази здатні розщеплювати ароматичні групи, що сприяє дегалогенуванню ароматичних сполук.
Прикладом видалення хлору є реакція дегідрохлорування інсектициду 2,2-біс(парахлорфеніл)-1,1,1-трихлоретану (ДДТ), яке відбувається дуже повільно з утворенням 2,2-біс(парахлорфеніл)-1,1- дихлоретилену (ДДЕ) (рис. 10.35).
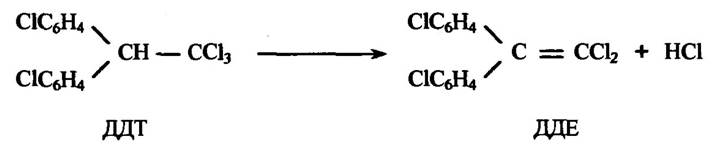
Рис. 10.35. Реакція дегідрохлорування ДДТ
Алкілгалогеніди (бромхлорметан, метилендихлорид, метилендибромід тощо) здатні до гідролітичного дегалогенування ферментами печінки та нирок з утворенням вільних галогенід-іонів (рис. 10.36).
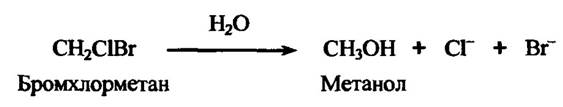
Рис. 10.36. Реакція гідролітичного дегалогенування бромхлорметану
Деякі ароматичні сполуки можуть утворювати феноли заміщенням галогену (рис. 10.37).
Аліфатичні та ароматичні галогеновані вуглеводи за взаємодії з глутаматом за участі ферментів печінки здатні перетворюватися в глутамінові похідні та меркаптурові кислоти заміщенням атома галогену.
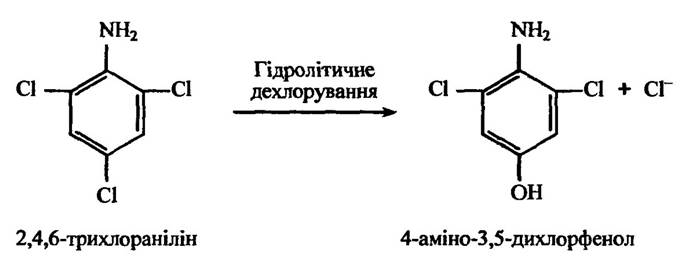
Рис. 10.37. Реакція гідролітичного дихлорування 2,4,6-трихлораніліну
Мікросомальними ферментами печінки за участі НАДФН і О2 відбувається відновне дегалогенування чотирихлористого карбону в хлороформ (рис. 10.38).

Рис. 10.38. Реакція відновного дегалогенування чотирихлористого карбону
Подібно відбувається дегалогенування анестезуючих речовин галотану, метоксифлюрану та деяких інших.
Поліхлорбіфеніли— це відносно стійкі токсичні сполуки. Відомо, що мікробна деградація біфенілу відбувається за участі систем реакцій катаболізму, які подібні до інших ароматичних вуглеводів.
Бензипірен — стійка поліароматична сполука, за деградації якої утворюються канцерогенні гідрокси- та епоксипохідні. Він не мінералізується в системах активного мулу, хоча описано кілька культур мікроорганізмів, які частково розщеплюють цю сполуку за допомогою гідроксилазних систем.
Стічні води нафтової промисловості очищують біологічним методом після вилучення більшої частини нафти коагулянтами. Ступінь мікробіологічної деградації нафтових розливів залежить від низки факторів, основним з яких є склад нафти, наявність харчових речовин для мікроорганізмів, зокрема нітрогену й фосфору, тощо.
Синтетичні поверхнево-активні речовини, які використовуються в промисловості та побуті, теж здатні до біодеградації. їх розклад починається, як правило, з окиснення кінцевих метальних груп, після чого за рахунок β-окиснення відбувається розщеплення лінійних бічних ланцюгів. Молекули, структура яких містить кільце, розкладаються, як правило, після цілковитої деградації бічного ланцюга.
Реакції кон’югації. Крім розглянутих реакцій окиснення, відновлення та деградації ксенобіотиків у їхній біотрансформації виділяють також, як уже зазначалося, реакції кон’югації. Вони становлять собою реакції біосинтезу, за якого ксенобіотики або їхні метаболіти з’єднуються з ендогенними сполуками (глюкуроновою кислотою, сульфатом, ацетил-КоА, гліцином, деякими ароматичними та аліфатичними амінами тощо) з утворенням кон’югатів. У цьому процесі ендогенні сполуки приєднуються до функціональних груп ксенобіотиків (гідроксильної, амінної, карбоксильної, епоксидної, атома галогену та ін.). Утворені в результаті кон’югації сполуки стають, як правило, полярнішими, менш ліпідорозчинними, що надає їм здатності легше виводитися з організмів.
У багатьох реакціях кон’югації ендогенні сполуки переносяться за участю коферментів. Серед коферментів у таких процесах широко використовуються уридиндифосфатні, зокрема уридиндифосфат-а- D-глюкоза (УДФГ) за утворення β-глюкозидів та уридиндифосфат-α-D-глюкуронова кислота (УДФГК) за утворення β-глукоронідів.
Глюкозидні кон’югати утворюються в комах, молюсків, рослин та бактерій перенесенням глюкози до ксенобітика від УДФГ.
У разі фенолів формування кон’югатів відбувається так (рис. 10.39).
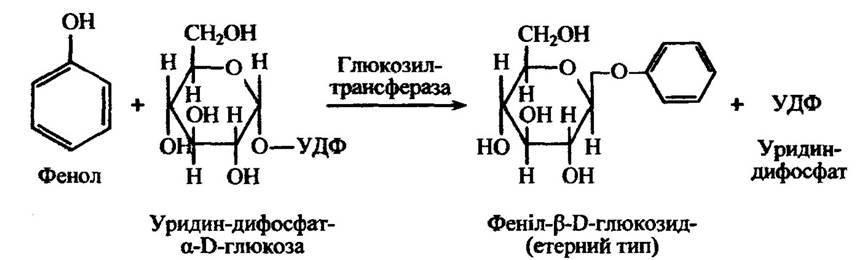
Рис. 10.39. Реакція утворення феніл-β-D-глюкозиду
У комах утворення глюкозидних кон’югатів є основним механізмом дезінтоксикації. Подібно до комах, у молюсків теж синтезуються глюкозиди.
У рослинах глюкозиди утворюються з природних сполук, які синтезуються самими рослинами, таких як алкалоїди, а також за дезінтоксикації чужорідних сполук.
Синтезуються, як правило, етерні та складноетерні глюкозиди. Можливе також утворення амігдалозидів. Останні утворюються як глюкозиди під час перенесення глюкози від УДФГ, за винятком того, що в цьому разі переносяться дві молекули глюкози. У бактерій теж синтезуються глюкозиди як фенолів, так і кислот, за участю УДФГ.
У печінці ссавців міститься УДФГ, але брак глюкозилтрансфераз перешкоджає утворенню глюкозидів.
У ссавців, птахів, рептилій і амфібій, але не в риб, утворюються глюкоронідні кон’югати.
Реакцію формування глюкоронідного кон’югату з фенолом наведено на рис. 10.40.
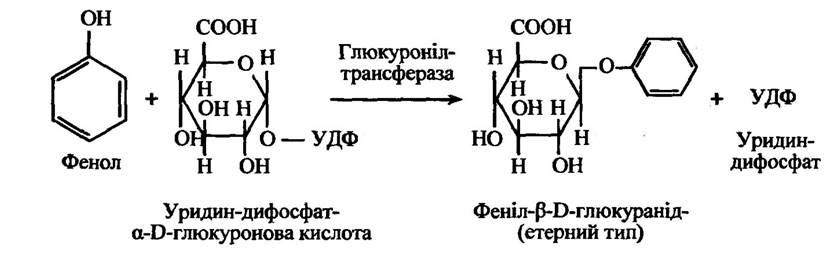
Рис. 10.40. Реакція утворення феніл-β-D-глюкуроніду
Такі ссавці, як кішки, характеризуються винятковим способом утворення глюкуронідних кон’югатів з фенолом через брак відповідних трансглюкуронілаз. Тому ці тварини особливо чутливі до токсичної дії фенолів. Однак у кішок виділяються глюкуроніди тироксину, білірубіну та іопанової кислоти в жовч, а також глюкуронідні кон’югати гідроксильованих метаболітів ацетамідофлюорену із сечею. Водночас риби мають необхідні глюкуронілтрансферази, але в них немає коферменту УДФГК.
Глюкуронідні кон’югати класифікуються на N-глюкуроніди, О- глюкуроніди та S-глюкуроніди. У свою чергу, О-глюкуроніди, які утворюються з фенолів, спиртів (у тому числі стероїди) і карбонових кислот, поділяють на чотири типи: 1)етерного типу (утворюються з фенолів і спиртів); 2) складноетерного типу (з карбонових кислот); 3)енольного типу (з псевдокислот, наприклад, 4-оксикумарину); 4) гідроксиламінового типу (зі сполук, які гідроксилюють- ся за амінною групою, наприклад, 2-ацетиламінофлюорен).
Відомо також кілька типів N-глюкуронідів. Так, атом нітрогену, до якого приєднується глюкуронідна частина, може бути в сульфамідній групі, аміногрупі, карбамільній групі тощо.
За кон’югації тіолових сполук (тіофелену, 2-меркаптобензолу та ін.) з глюкуроновою кислотою утворюються S-глюкуроніди.
Глікозиди (глюкозиди та глюкуроніди) зазвичай полярніші, ніж вихідні сполуки, та виводяться з місця утворення у тварин з виділеннями, а в рослинах — шляхом концентрування у вакуолях клітин листя та інших тканин.
Важливим типом кон’югантів є складні етери сірчаної кислоти — етерсульфати. Вони утворюються у ссавців, птахів, рептилій, амфібій, але не в риб. Серед безхребетних етерсульфати формуються в членистоногих, комах і, можливо, у деяких видів молюсків.
Етерсульфати поділяють на кілька типів: 1) арилсульфати — складні етери фенолових сполук; 2) алкілсульфати — складні етери первинних аліфатичних спиртів; 3) сульфамати — складні етери сірчаної кислоти та амінів, які містять сульфамідну групу; 4) стероїдні сульфати — складні етери спиртових груп стероїдного бічного ланцюга, складні етери циклічних бокових груп, складні етери фенолових стероїдів; 5) вуглеводні сульфамати — складні етери гідроксильних груп вуглеводів.
Серед цих п’яти груп найпоширенішими типами етерсульфатів є арилсульфати, алкілсульфати.
Етерсульфати синтезуються перенесенням сульфату від аденозин-3'-фосфат-5'-фосфосульфату (ФАФС) до фенолів, спиртів або амінів відповідними сульфотрансферазами. Утворюється ФАФС з аденозин-5 -трифосфату (АТФ) та аденозин-5 '-фосфосульфату (АФС) (рис. 10.41).
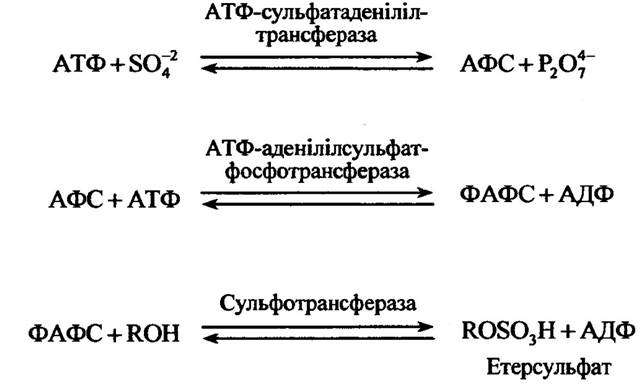
Рис. 10.41. Реакції утворення етерсульфатів
У багатьох тварин і деяких мікроорганізмів відбувається утворення N-, О- і S-метилових кон’югатів під час перенесення метальної групи від універсального донора метальних груп S-аденозилметіоніну в процесах трансметилювання, який синтезується з метіоніну в реакції з АТФ, на феноли, аміни та тіолові сполуки. Реакцію утворення S-аденозилметіоніну ілюструє рис. 10.42.
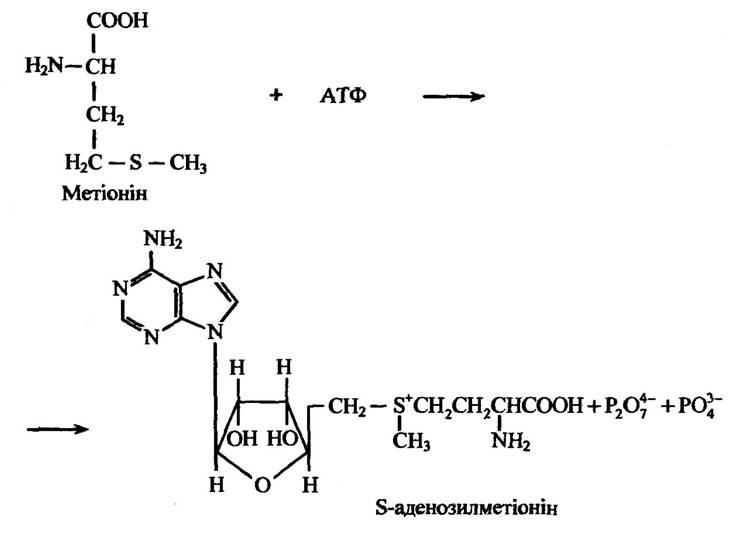
Рис. 10.42. Реакція утворення S-аденозилметіоніну
З етилового аналогу метіоніну (етиніну) синтезується аналогічно S-аденозилетинін, який бере участь у реакціях трансетилювання.
У більшості ссавців, деяких птахів, земноводних і комах відбувається утворення N-метилових кон’югатів зі сполуками, які притаманні організмам та ксенобіотикам. Так, наприклад, фенілетаноламін-N-метилтрансфераза каталізує синтез адреналіну з норадреналіну (рис. 10.43), N-метилювання інших фенілетаноламінових похідних.

Рис. 10.43. Реакція утворення адреналіну за метилювання норадреналіну
Неспецифічна N-метилтрансфераза метилює ендогенні та чужорідні первинні та вторинні аміни (серотонін, триптамін, норникотин, нормепірин, піридин тощо). Реакції метилювання серотоніну та піридину наведено на рис. 10.44.
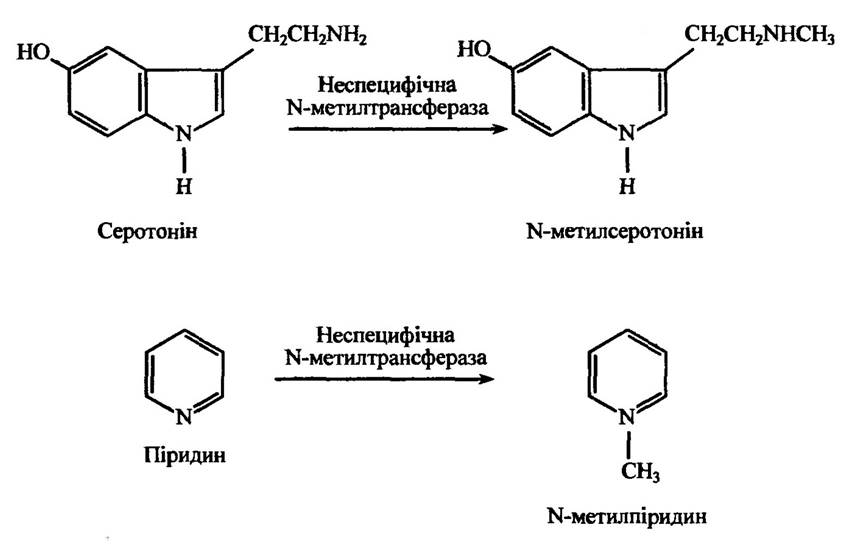
Рис. 10.44. Реакції утворення N-метилсеротоніну і N-метилпіридину
за метилювання серотоніну та піридину відповідно
Відомі також специфічні N-метилтрансферази, які метилюють ендогенні сполуки — нікотинамід, пуринові та піримідинові поліну- клеотиди, фосфатидиламіноетанол тощо. Реакція метилювання гістаміну імідазол-М-метилтрансферазою ілюструє рис. 10.45.
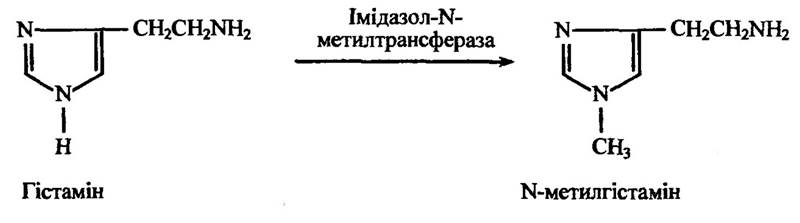
Рис. 10.45. Реакція утворення N-меггилгістаміну за метилювання гістаміну
Відомі також специфічні N-метилтрансферази ендогенних сполук, які N-метилюють нікотинамід, гуанідоцтову кислоту, фосфати- диламіноетанол, пуринові та піримідинові полінуклеотиди.
Катехоламінові гормони, а також ряд катехоламінових ксенобіоти- ків, зокрема галова (3,4,5-триоксибензойна) та кофеїнова (3,4-діоксикорична) кислоти метилюються катехол-О-метилтрансферазою (рис. 10.46). Цей фермент міститься в печінці, нирках, крові, шкірі, нервових волокнах, тканині залоз. Як метильний донор використовується S-аденозилметіонін.
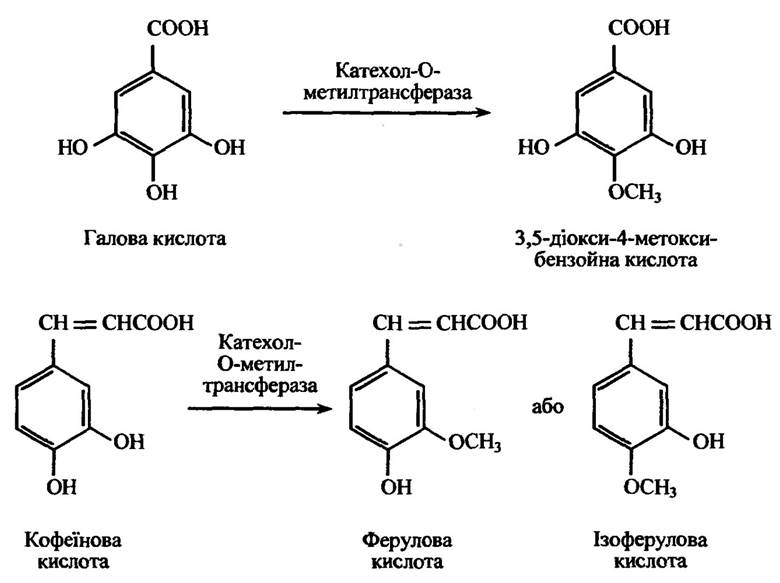
Рис. 10.46. Реакція метилювання похідних катехоламінів, яка визначається положенням інших замісників в ароматичному кільці
Оксиіндол-О-метилтрансфераза метилює N-ацетилсеротонін (рис. 10.47), буфеїн та інші оксиіндоли. Цей фермент у хребетних виявлено в гіпофізі та сітківці ока. Його відмінність від катехол-О-метилтрансферази полягає в тому, що він не метилює катехоли та для jioro функціонування не потрібні іони магнію.
Йодофенол-О-метилтрансфераза метилює ортойодфеноли, зокрема 3,5-дийод-4-оксибензойну кислоту (рис. 10.48), але не тироксин або трийодтироксин.
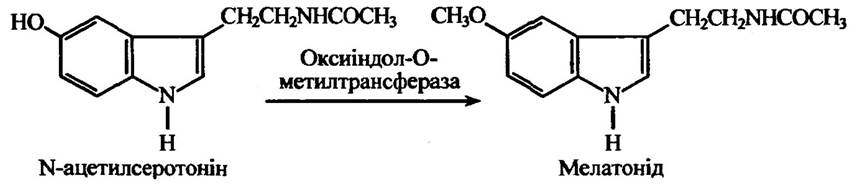
Рис. 10.47. Реакція метилювання оксиіндол-О-метилтрансферазою N-ацетилсеротоніну
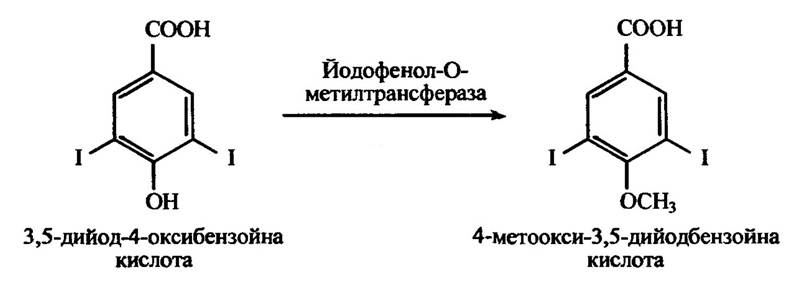
Рис. 10.48. Реакція метилювання йодофенол-О-метилтрансферазою 3,5-дийод-4-оксибензойної кислоти
У щурів, собак та, імовірно, деяких інших ссавців метилюється морфін з утворенням кодеїну шляхом кон’югації, і для реакції S- аденозилметіонін як донор метильних груп не потрібен.
Метильні групи можуть переноситися також до тіолових груп ксенобіотиків (метилмеркаптанів, етилмеркаптанів, меркаптооцтової кислоти, меркапетанолу, димеркапторпанолу, тіерацилу тощо) у реакціях S-метилювання в печінці, нирках, легенях. Такі ендогенні тіолові сполуки, як цистеїн, гомоцистеїн і глютатіон, не метилюються цим ферментом. Реакцію S-метилювання наведено на рис. 10.49.

Рис. 10.49. Реакція метилювання етантіолу S-метилтрансферазою
Ароматичні аміносполуки та сульфаміди здатні ацетилюватися. Існують певні відмінності перебігу цих реакцій у деяких ссавців. Так, у собак і лисиць виділяється значна кількість ароматичних амінів у вигляді ацетильованих похідних через наявність у них інгібітору ариламінацетилтрансферази в печінці та нирках, або високої активності ароматичної дезацилази. Водночас у цих тварин аліфатичні аміногрупи легко ацетилюються.
У дорослих птахів ароматичні аміни ацетилюються, але в курчат через наявність у нирках великої кількості ароматичної дезацилази це не відбувається. У деяких земноводних і риб також можуть ацетилюватись ароматичні аміни, тимчасом як у рептилій такий тип кон’югації не спостерігається. Відомо, що в деяких видів комах (сарани, молі тощо) відбувається ацетилювання ароматичних амінів, а також їх дезацетилювання. Аналогічне ацетилювання та дезацетилювання здійснюється в рослинах.
Чужорідні аміни можуть ацетилюватися через проміжні сполуки з коензимом A (KoA-SH) з утворенням амідних кон’югатів (рис. 10.50).
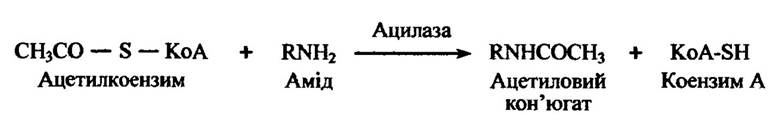
Рис. 10.50. Реакція ацетилювання амідів ацетилкоензимом А
Ацетилюватися здатні ароматичні аміни, сульфаміди та деякі чужорідні ароматичні амінокислоти (рис. 10.51).
Рис. 10.51. Реакція ацетилювання аніліну, S-фенілцистеїну та ізоціаніду
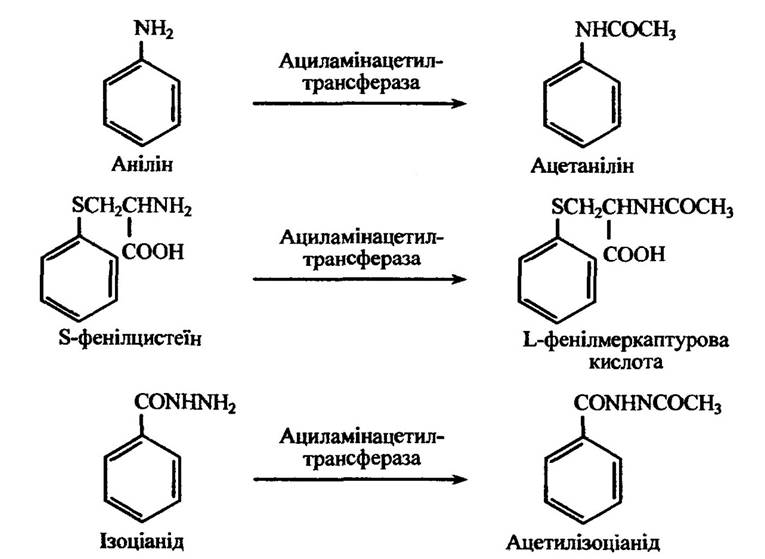
Рис. 10.51. Реакції ацетилювання аніліну, S-фенілцистеїну та ізоціаніду
Ацетилювання відбувається, як правило, у печінці, але можливе також у ретикулоендотеліальних клітинах селезінки та легенів, у слизовій шлунково-кишкового тракту.
Характерною реакцією ароматичних карбонових кислот є кон’югація з гліцином (утворення так званих гілурових кислот) та іншими амінокислотами. Заміщені оцтові кислоти (фенілоцтова та індолілоцтова), акрилові (корична) та деякі природні стероїдні кислоти (холева кислота) теж утворюють гліцинові кислоти. Водночас аліфатичні карбонові кислоти їх не утворюють.
У загальному вигляді реакції пептидної кон’югації з гліцином ілюструє рис. 10.52.
У разі утворення гліцинових кон’югатів з бензойною кислотою відбуваються такі реакції (рис. 10.53).
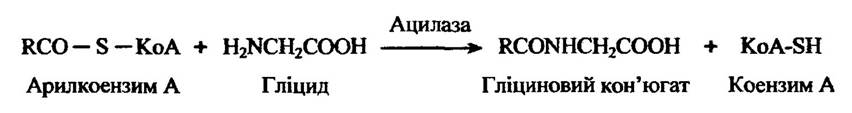
Рис. 10.52. Реакції пептидної кон’югації з гліцином
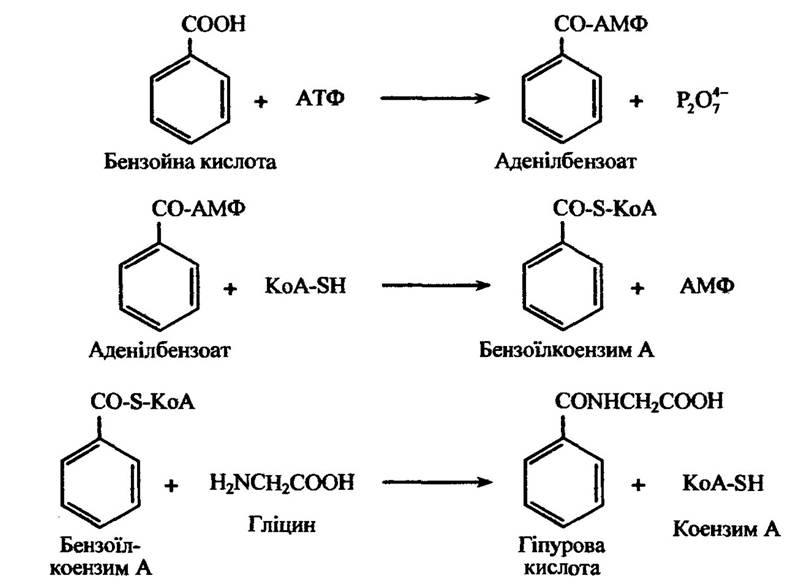
Рис. 10.53. Реакції утворення гліцинових кон’югатів з бензойною кислотою
Гліцин є амінокислотою, яка найчастіше бере участь у пептидній кон’югації у ссавців, але в людини й деяких приматів утворення кон’югатів відбувається за допомогою фенілоцтової кислоти: заміщені фенілоцтові кислоти формують гліцинові кон’югати. Крім того, у людини та інших приматів індол-3-оцтова кислота утворює глутамі-новий кон’югат, а n-аміносаліцилова кислота кон’югує з глутаміном.
В інших тварин пептидна кон’югація відбувається за участю інших амінокислот. Так, наприклад, у рептилій і деяких птахів у пептидній кон’югації бере участь орнітин, у деяких павукоподібних і багатоніжок — аргінін і глутамін. У деяких приматів також кон’югати утворюються з глутаміном. У кішок з сечею виділяється хінальдинова кислота (хінолін-2-карбонова кислота) у вигляді кон’югатів хінальдилгліцилтаурину і хінальдилгліцилгліцину (рис. 10.54).
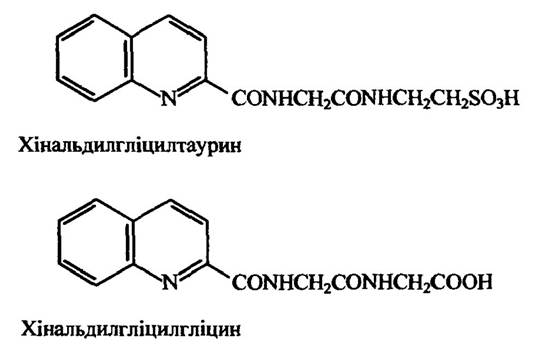
Рис. 10.54. Продукти кон’югації з хінальдиновою кислотою
Вибір амінокислоти, яка бере участь у кон’югації, визначається її значенням у метаболізмі відповідного виду.
Ароматичні та аліфатичні ксенобіотики здатні також кон’югувати з глутатіоном (G-SH), утворюючи S-алкілглутатіони і S-арил- глутатіони, похідні цистеїну і, зрештою, меркаптурові кислоти. Серед різноманітних типів такої кон’югації найпоширенішими є глутатіонові кон’югати ароматичних вуглеводів і такі, в яких сполуки з глутатіоном кон’югують у результаті заміщення атомів галогену, нітроамідних і сульфамідних груп.
У більшості ссавців і комах відбувається спочатку кон’югація з глутатіоном і подальшим перетворенням цього кон’югату у відповідне цистеїнове похідне, а відтак ацетилювання з утворенням премер- каптурової кислоти. Проте в деяких ссавців, наприклад мурчаків, меркаптурові похідні майже не утворюються через нестачу ферменту, який ацетилює арилцистеїни. У сарани теж виділяються неаце- тильовані цистеїнові похідні.
У процесі утворення глутатіонових кон’югатів таких ароматичних вуглеводів, як бензол, нафталін, антрацен тощо, ці сполуки перетворюються в меркаптуронові кислоти, в яких атом гідрогену заміщений L-ацетилцистеїновою ланкою. Ці кон’югати виділяються із сечею у вигляді премеркаптурових кислот. Вони під час оброблення мінеральними кислотами утворюють меркаптурову кислоту (відщеплюється молекула води) та феноли (відщеплюється N-ацетилцистеїн). Реакцію утворення глутатіонового кон’югату бензолу наведено на рис. 10.55.
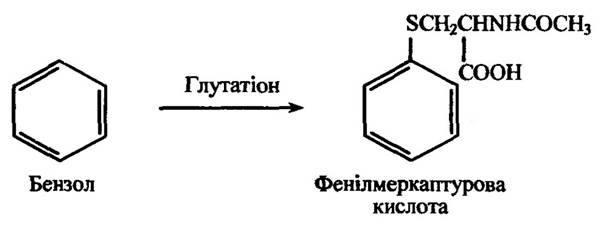
Рис. 10.55. Реакція кон’югації бензолу за участю глутатіону
Ряд сполук, зокрема бензилхлорид, 4-хлорнітробензол, 2,4-дихлор-нітробензол, 1- і 2-менафтилхлориди та інші, з лабільним атомом хлору утворюють меркаптурові кислоти, в яких L-ацетилцистеїновий ланцюг заміщує хлор. Таку реакцію з бензилхлоридом наведено на рис. 10.56.
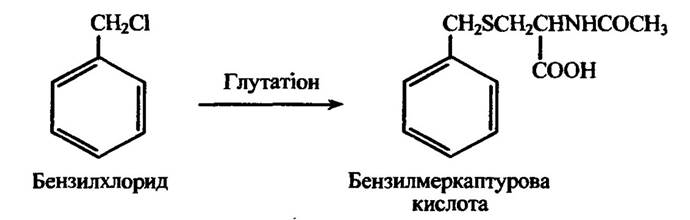
Рис. 10.56. Реакція кон’югації бензилхлориду за участю глутатіону
Ряд галагеноалкан і в і нітроалканів також утворюють меркаптурові кислоти заміщенням атомів галогену або нітрогруп за механізмом, очевидно, аналогічним синтезу бензилмеркаптурової кислоти. Метаболітами бромалканів можуть бути алкілмеркаптурові або оксиалкілмеркаптурові кислоти (рис. 10.57).
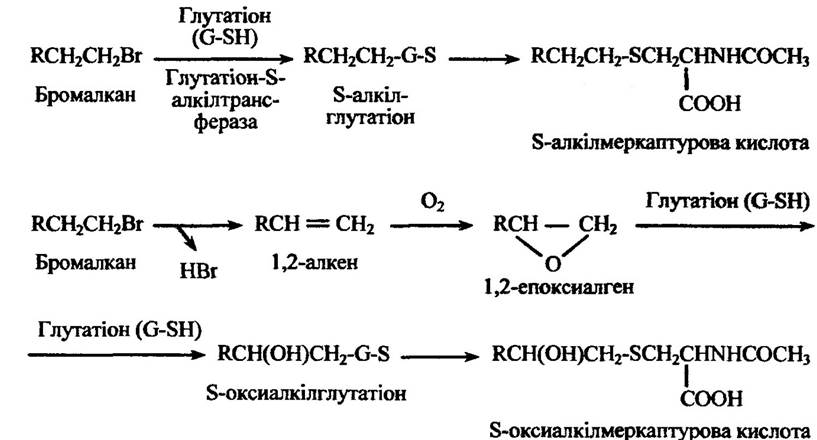
Рис. 10.57. Реакції кон’югації бромалкану з утворенням алкілмеркаптурової
або оксиалкілмеркаптурової кислот
У хребетних (у тому числі людини), безхребетних, рослин та бактерій детоксикація ціанідів може відбуватися шляхом їх кон’югації з сульфуром (рис. 10.58) з утворенням тіоциану (роданідів). Фермені риданаза, який каталізує цю кон’югацію з хребетних, локалізований у всіх тканинах (за винятком крові), і особливо в печінці та нирках. У цій реакції тіосульфат, цистеїн і глутатіон не виступають у ролі донорів сульфуру.
![]()
Рис. 10.58. Реакції кон’югації ціанідів за участю родонази
Ціанід може дезінтоксикуватися також у реакції з цистином, утворюючи 2-амінотіазолідин-4-карбонову кислоту (рис. 10.59).
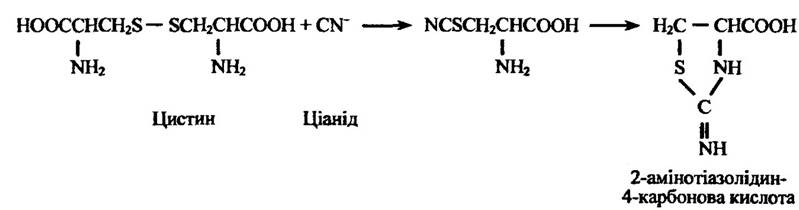
Рис. 10.59. Схема кон’югації цистину з ціанідом
Якщо молекула ксенобіотика має дві або більше функціональних груп, то зазвичай кон’югує тільки одна група. Разом з тим можуть утворюватися і подвійні кон’югати. Так, наприклад, ацетамідофеноли та оксианізоли здатні знову кон’югувати з глюкуроновою кислотою або сульфатом.
Наведений перелік реакцій кон’югації далеко не вичерпує всіх можливостей. Так, наприклад, існує ще фосфатна кон’югація, з гліцинтаурином, формілом тощо. З легніном у рослин можуть кон’югувати пестициди 2,4-дихлорфеноксиоцтової кислоти, 3,4-дихлоранілу, пентахлорфенолу. Іони важких металів кон’югують із металотіонінами.
Зауважимо, що кон’югація ксенобіотиків як знешкодження їхньої негативної дії характерна тільки для певного організму. Ксенобіотики, що потрапляють в екосистему, продовжують у трансформованій формі циркулювати в ній.