ОСНОВЫ БИОХИМИИ ЛЕНИНДЖЕРА - ТОМ 1. ОСНОВЫ БИОХИМИИ СТРОЕНИЕ И КАТАЛИЗ - 2011
ЧАСТЬ I. СТРОЕНИЕ И КАТАЛИЗ
3. АМИНОКИСЛОТЫ, ПЕПТИДЫ И БЕЛКИ
3.4. Структура белка: первичная структура
Очистка белка - это лишь начало детального биохимического анализа его структуры и функций. Почему один белок является ферментом, другой — гормоном, третий — структурным белком, а еще один — антителом? Чем они принципиально различаются с химической точки зрения? Наиболее очевидные различия проявляются в структуре белков на каждом уровне их организации.
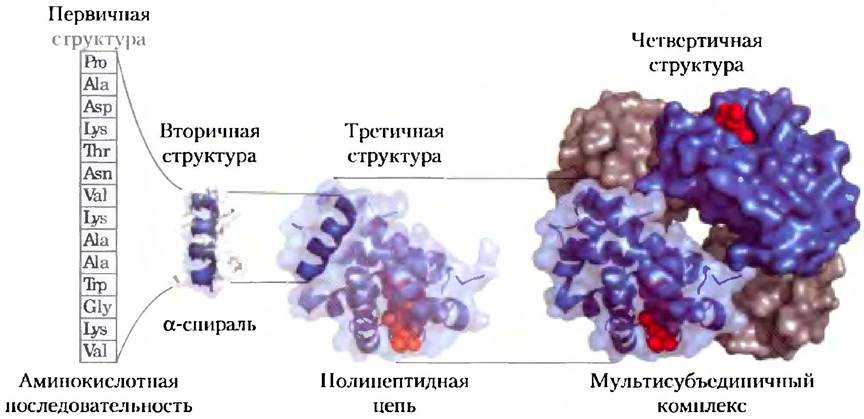
Задача описания и понимания структуры таких крупных макромолекул, как белки, решается на разных уровнях сложности. Обычно для белков выделяют четыре уровня структурной организации (рис. 3-23). Все ковалентные связи (главным образом пептидные связи и дисульфидные мостики), соединяющие аминокислотные остатки в полипептидную цепь, представляют собой первичную структуру белка. Наиболее важным элементом первичной структуры является последовательность аминокислотных остатков. Вторичной структурой называют некоторые наиболее часто встречающиеся способы укладки аминокислотной цепи, формирующие характерные структурные элементы. Третичная структура описывает все аспекты пространственной организации белка. Если белок состоит из двух или нескольких полипептидных цепей, их пространственное взаиморасположение описывается четвертичной структурой. При знакомстве с белками мы рассмотрим даже такие сложные белковые агрегаты, которые состоят из десятков и даже тысяч субъединиц. Первичная структура белка является предметом рассмотрения в настоящей главе; более высокие уровни организации белков обсуждаются в гл. 4.
Рис. 3-23. Уровни организации белковых молекул. Первичная структура белка представляет собой последовательность аминокислот, связанных между собой пептидными связями и дисульфидными мостиками. Образующийся полипептид может сворачиваться в определенную вторичную структуру, например, в α-спираль. Спираль является частью третичной структуры молекулы, которая, в свою очередь, может быть лишь одной из нескольких субъединиц мультисубъединичного белка, вместе формирующих его четвертичную структуру (на рисунке изображена структура гемоглобина).
Особенно информативными могут быть различия в первичной структуре. Каждый белок характеризуется определенным числом и последовательностью аминокислотных остатков. Как мы увидим в гл. 4, именно первичная структура белка определяет его трехмерную структуру, которая в свою очередь отвечает за функции белка. В фокусе нашего внимания в данной главе будет первичная структура белка. Прежде всего, мы остановимся на эмпирическом правиле, что аминокислотная последовательность белка и его функции тесно связаны между собой. Затем мы рассмотрим способы определения аминокислотной последовательности белка и в завершение поговорим о том, как много информации можно почерпнуть из первичной структуры белка.
Функция белка зависит от аминокислотной последовательности
Бактерия Escherichia coli синтезирует свыше 3000 различных белков, а человек имеет около 25 000 генов, кодирующих еще большее количество белков (генетические аспекты обсуждаются в части III настоящей книги). В обоих случаях каждый тип белка характеризуется уникальной трехмерной структурой, необходимой для выполнения определенной функции. Каждый белок, кроме того, имеет уникальную аминокислотную последовательность. Интуиция подсказывает, что именно аминокислотная последовательность должна определять трехмерную структуру белка и, в конечном итоге, его функции. Но так ли это на самом деле? Беглый обзор многообразия первичных структур белков дает ряд эмпирических доказательств, позволяющих утверждать наличие тесной связи между аминокислотной последовательностью и биологической функцией.
Прежде всего, как мы отмечали выше, белки с различными функциями всегда различаются по аминокислотной последовательности. Кроме того, тысячи различных генетических заболеваний человека сопровождаются образованием аномальных белков. Дефекты могут быть различными: от замены одной-единственной аминокислоты (как при серповидноклеточной анемии, см. гл. 5) до деления большого участка полипептидной цепи (как в большинстве случаев мышечной дистрофии Дюшенна, при которой делеция большого участка гена, кодирующего белок дистрофии, приводит к синтезу короткого неактивного белка). Теперь мы знаем, что если изменена первичная последовательность, то и функция белка может измениться. Наконец, при сравнении сходных по функциям белков у организмов, относящихся к разным видам, часто выясняется, что эти белки имеют много общего в аминокислотных последовательностях. Например, белок убиквитин, состоящий из 76 аминокислотных остатков, участвует в регуляции деградации других белков. Было показано, что его последовательность полностью идентична у таких несхожих видов, как фруктовая мушка и человек.
Является ли аминокислотная последовательность определенного белка абсолютно инвариантной? Ответ такой: нет, возможны некоторые отклонения. По оценкам, 20-30% белков человека полиморфны, т. е. в популяции людей эти белки имеют несколько различающиеся аминокислотные последовательности. Многие из этих вариаций последовательности практически нс оказывают влияния на функционирование белка. Более того, белки со схожими функциями из отдаленно родственных видов могут сильно различаться по размеру и аминокислотной последовательности.
Изменения в некоторых участках аминокислотной последовательности могут не повлиять на функции белка, однако в большинстве белков существуют участки последовательности, особенно важные для их активности, и эти участки консервативны. Доля последовательности, наиболее важной для функционирования различных белков, разная, что усложняет задачу сопоставления первичной и третичной структур, а также структуры и функций. Прежде чем подробнее остановиться на этой проблеме, мы поговорим о том, как определяют аминокислотную последовательность белков.
Уже расшифрованы аминокислотные последовательности миллионов белков
Два больших открытия, сделанные в 1953 г., сыграли важнейшую роль в истории биохимии. В 1953 г. Джеймс Д. Уотсон и Фрэнсис Крик создали модель двойной спирали ДНК и предсказали структурный механизм ее репликации (гл. 8). Их предположение раскрыло молекулярные основы идеи гена. Тогда же Фредерик Сейгер определил аминокислотную последовательность гормона инсулина (рис. 3-24), удивив многих ученых, считавших расшифровку аминокислотной последовательности полипептидной цепи безнадежно трудным делом. Скоро стало понятно, что нуклеотидная последовательность ДНК и аминокислотная последовательность белка каким-то образом связаны между собой. Всего через десять лет после этих открытий было установлено, что именно ДНК определяет аминокислотную последовательность белка (гл. 27). Сегодня огромное число белковых последовательностей можно определить на основании последовательностей ДНК, собранных в постоянно растущих банках данных. Однако в современной химии белка часто пользуются традиционным методом секвенирования белков, поскольку так иногда удастся выявить детали, которые нельзя установить на основании последовательности гена, такие как модификации, происходящие после завершения синтеза белка.
Рис. 3-24. Аминокислотная последовательность инсулина быка. Две полипептидные цепи белка соединены между собой дисульфидными мостиками. Инсулины человека, свиньи, собаки, кролика и кита имеют одинаковые А-цепи, а инсулины коровы, свиньи, собаки, козы и лошади имеют идентичные В-цепи.

Химический метод секвенирования белка сегодня дополняет все удлиняющийся список новых методов, обеспечивающих многочисленные пути получения такого рода данных, которые играют важнейшую роль во всех областях биохимических исследований.
Фредерик Сенгер

Короткие полипептиды секвенируют с помощью автоматических секвенаторов
Для анализа первичной структуры белка применяют различные методы. Разработаны методики, позволяющие пометить и идентифицировать N-концевой аминокислотный остаток (рис. 3-25, а). Сенгер для этой цели предложил использовать 1-фтор-2,4-динитробензол. Для этой же цели применяют дансилхлорид и дабсилхлорид, производные которых легче детектируются, чем динитрофенильные производные. После связывания одного из этих реагентов с N-концевым остатком осуществляют гидролиз полипептидной цепи (6 М НСl) до составляющих ее отдельных аминокислот и определяют меченый остаток. Поскольку в результате данной процедуры полипептид разрушается, этот метод нельзя использовать для определения других остатков, за исключением концевого. Однако таким образом можно определить число отдельных полипептидных цепей в белке, если все они имеют различные N-концы. Например, если подвергнуть такой процедуре инсулин, то можно определить два N-концевых остатка — Рhе и Сlу (рис. 3-24).
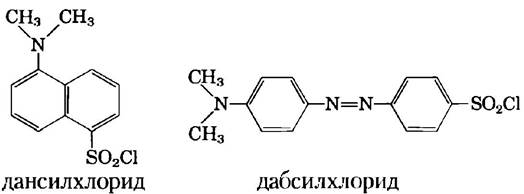
Рис. 3-25. Стадии секвенирования полипептидной цепи, а) Первой стадией секвестрования может быть определение N-концевого аминокислотного остатка. Здесь показана схема определения N-концевого остатка по методу Сенгера.) Метод Эдмана позволяет определить полную аминокислотную последовательность. В случае коротких пептидов этот метод позволяет полностью определить всю последовательность, и тогда стадию (а) часто пропускают. Стадия (а) полезна в случае больших белков, которые перед секвенированием предварительно разделяют на более мелкие фрагменты (рис. 3-27).
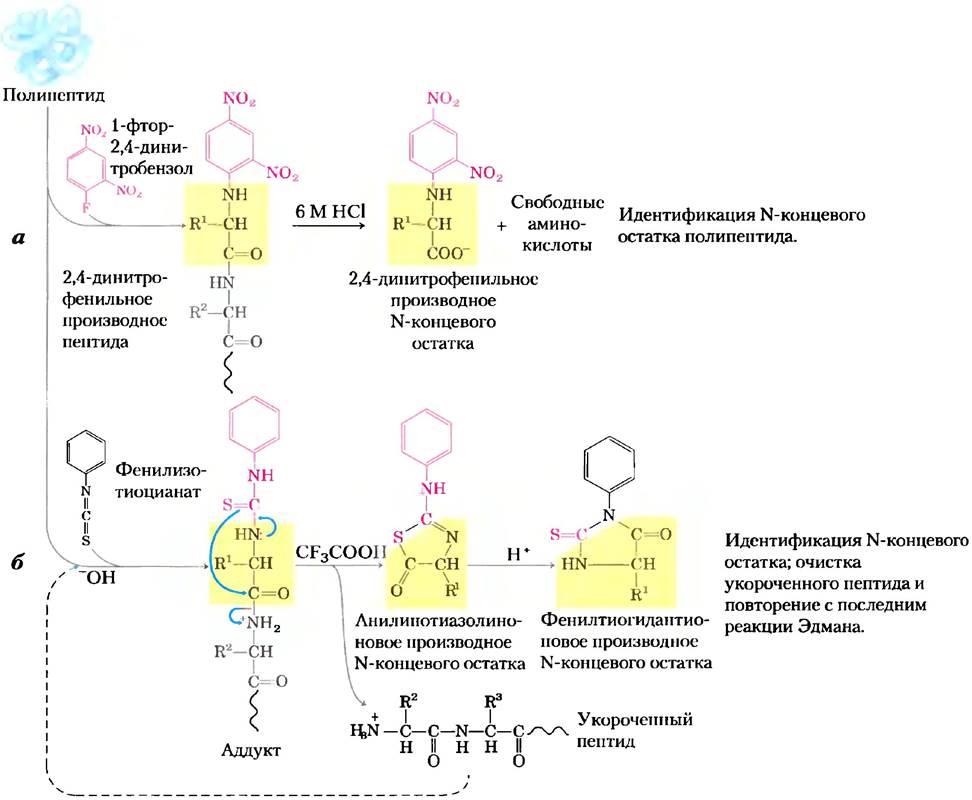
Чтобы ссквенированать (от англ. sequence — последовательность) весь белок, обычно применяют метод, предложенный Пером Эдманом. В методе деградации по Эдмануудается отделить от полипептидной цепи только N-концевой остаток, не затрагивая все остальные (рис. 3-25, б). Для этого в слабощелочных условиях пептид обрабатывают фенилизотиоцианатом, который присоединяется к N-концевой аминокислоте. Затем образовавшийся аддукт отщепляют с помощью безводной трифторуксусной кислоты в виде анилинотиазолинонового производного, экстрагируют органическими растворителями, в водном растворе кислоты превращают в более устойчивое фенилтиогидантионовое производное и идентифицируют. Последовательное проведение реакций сначала в щелочных, а затем в кислых условиях позволяет контролировать ход всего процесса. Все реакции, затрагивающие N-концевой аминокислотный остаток, никак не влияют на остальную аминокислотную последовательность. После удаления и идентификации концевого остатка можно пометить вновь образовавшийся N-концевой остаток, удалить и вновь идентифицировать с помощью той же последовательности реакций. Эту процедуру повторяют до тех пор, пока не определят все аминокислотные остатки пептидной цени. Определение аминокислотной последовательности по методу Эдмана осуществляют с помощью прибора секвенатора, в котором стадии смешивания реагентов, разделения продуктов, идентификации и записи результатов полностью автоматизированы. Данный метод необычайно чувствителен. Иногда с его помощью удается определить аминокислотную последовательность белка, имея в руках всего несколько микрограммов образца.
Длина полипептидной цепи, которую можно секвенировать по методу Эдмана, зависит от эффективности отдельных химических стадий. Представьте себе белок, на N-конце которого расположена последовательность Gly-Pro-Lys и т. д. Если глицин был удален с эффективностью 97%, это означает, что 3% полипептидных молекул в растворе по-прежнему имеют на N-конце остаток глицина. Во втором цикле пролин составляет 94% свободных аминокислот (97% остатков Pro отщепляются от 97% молекул, заканчивающихся на Pro), а глицин составляет 2,9% (97% остатков Gly отщепляются от 3% молекул, заканчивающихся на Gly), а 3% молекул сохранят на N-конце глицин (0,1 %) или пролин (2,9%). И так в каждом цикле те пептиды, которые не отщепились на предыдущей стадии, будут вносить все более и более возрастающую погрешность; в конечном итоге станет невозможно определить, какая же аминокислота находится в настоящий момент на конце цепи. В современных секвенаторах достигается эффективность, превышающая 99% за цикл, так что с их помощью можно определить последовательность из 50 и более остатков. Первичную структуру инсулина, над которой Сенгер с сотрудниками трудились более 10 лет, сегодня можно определить за 1-2 дня прямым секвенированием в белковом секвенаторе. (Как мы будем обсуждать в гл. 8, секвенирование ДНК даже более эффективно.)
Крупные белки перед секвенированием необходимо разделить на фрагменты
По мере увеличения длины полипептида число ошибок в определении аминокислотной последовательности обычно возрастает. Для определения последовательностей больших полипептидов и белков их необходимо разделить па более мелкие фрагменты, а затем каждый секвенировать отдельно. Данный процесс включает в себя несколько стадий. Сначала белок расщепляют на специфические фрагменты с помощью химических или ферментативных методов. Если в белке существуют дисульфидные связи, их следует разрушить. Каждый фрагмент выделяют из смеси и секвенируют по методу Эдмана. В заключение определяют порядок расположения фрагментов в исходной цепи и локализацию дисульфидных связей, если они есть.
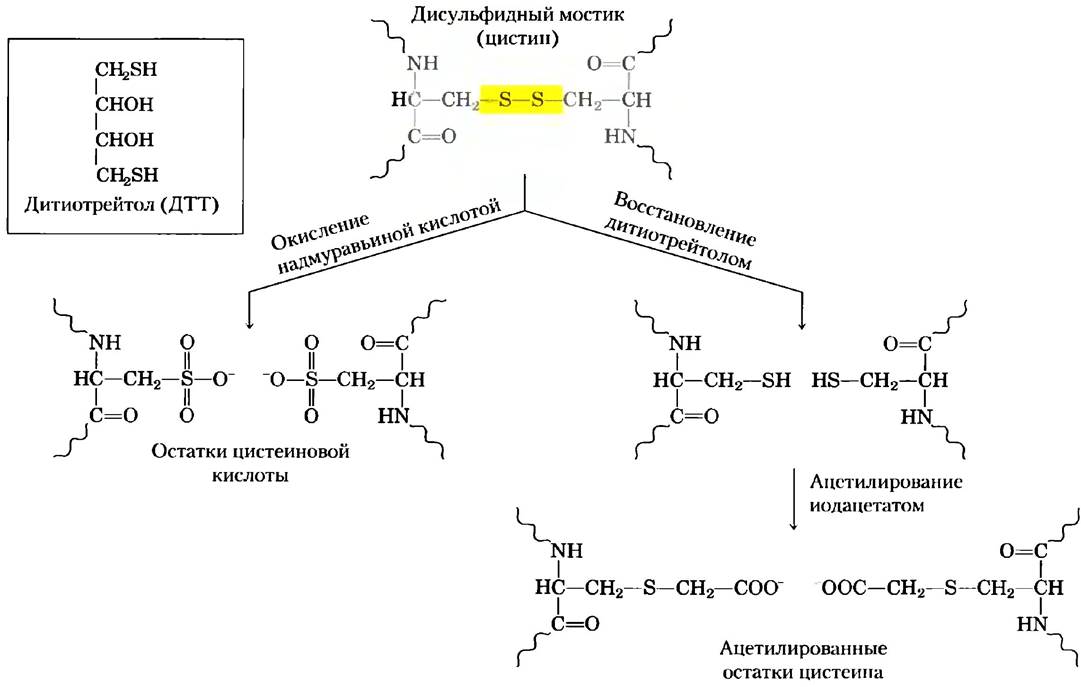
Разрыв дисульфидных связей. Наличие дисульфидных связей мешает определению аминокислотной последовательности. Остаток цистеина (рис. 3-7), отщепленный от полипептидной цени в реакции Эдмана, может остаться связанным с другой полипептидной цепью дисульфидным мостиком. Кроме того, дисульфидные мостики мешают ферментативному и химическому расщеплению полипептидной цепи на фрагменты. На рис. 3-26 схематично изображены два способа необратимого расщепления дисульфидных связей.
Рис. 3-26. Расщепление дисульфидных мостиков в белках двумя известными методами. В результате окисления остатков цистеина под действием надмуравьиной кислоты образуются остатки цистеиновой кислоты. После восстановления дитиотрейтолом или β-меркаптоэтанолом с образованием двух остатков цистеина необходимо провести дальнейшую модификацию реакционноспособных -SH групп, чтобы предотвратить появление нового дисульфидного мостика. Для этой цели используют ацетилирование с помощью иодацетата.
Расщепление полипептидной цепи. Существует несколько методов разделения полипептидной цепи на фрагменты. Гидролиз пептидных связей катализируют ферменты протеазы. Некоторые протеазы специфически расщепляют только тс пептидные связи, которые связывают определенные аминокислотные остатки (табл. 3-7). С помощью таких протеаз можно воспроизводимо получать предсказуемые фрагменты. Некоторые химические реагенты также способны расщеплять пептидную связь между определенными аминокислотами.
Таблица 3-7. Специфичность некоторых методов расщепления полипептидной цепи
Реагент (биологический источник)* |
Участок расщепления** |
Трипсин (поджелудочная быка) |
Lys. Arg (С) |
Протеаза из подчелюстной железы (мышь) |
Arg (С) |
Химотрипсин (поджелудочная быка) |
Phe, Тгр, Туг (С) |
Протеаза V8 (бактерия Staphylococcus aureus) |
Asp, Glu (С) |
Asp-N-протеаза (бактерия Pseudomonas fragi) |
Asp, Glu (N) |
Пепсин (желудок свиньи) |
Leu, Phe, Trp, Tyr (N) |
Эндопротеиназа Lys С (бактерия Lysobacter enzymogenes) |
Lys (С) |
Бромциан |
Met (С) |
* Все реагенты, за исключением бромциана, являются протеазами; все доступны из коммерческих источников.
** Указанные аминокислотные остатки являются участками узнавания фермента или реагента, которые расщепляют пептидную связь на С- или N-стороне указанного остатка.
Пищеварительный фермент трипсин, относящийся к классу протеаз, катализирует гидролиз только тех пептидных связей, карбонильная группа которых принадлежит остатку лизина или аргинина, вне зависимости от длины полипептидной цепи. Таким образом, зная общее число остатков Lys и Arg в полипептидной цепи, определенное из реакции полного гидролиза образца (рис. 3-27), можно предсказать число фрагментов, на которые расщепится полипептидная цепь при обработке трипсином. Полипептид, имеющий в последовательности пять остатков Lys и (или) Arg, в результате действия трипсина должен расщепиться на шесть фрагментов. Каждый из этих фрагментов, за исключением одного, будет иметь на С-конце остаток Lys или Arg. Фрагменты, полученные в результате расщепления трипсином, другим ферментом или химическим реагентом, далее подвергают очистке хроматографическим или электрофоретическим методом.
Рис. 3-27. Расщепление и секвенирование пептидов и определение порядка расположения пептидных фрагментов. В первую очередь определяют аминокислотный состав и N-концевую аминокислоту в исходном пептиде. Затем разрушают все имеющиеся в молекуле дисульфидные связи, мешающие секвенированию. В данном полипептиде есть только два остатка цистеина (С) и, соответственно, только одно возможное место локализации дисульфидной связи. В полипептидах с тремя и большим числом остатков цистеина расположение дисульфидных мостиков определяют так, как это описано в тексте. Расшифровку однобуквенных и трехбуквенных обозначений аминокислот см. в табл. 3-1.
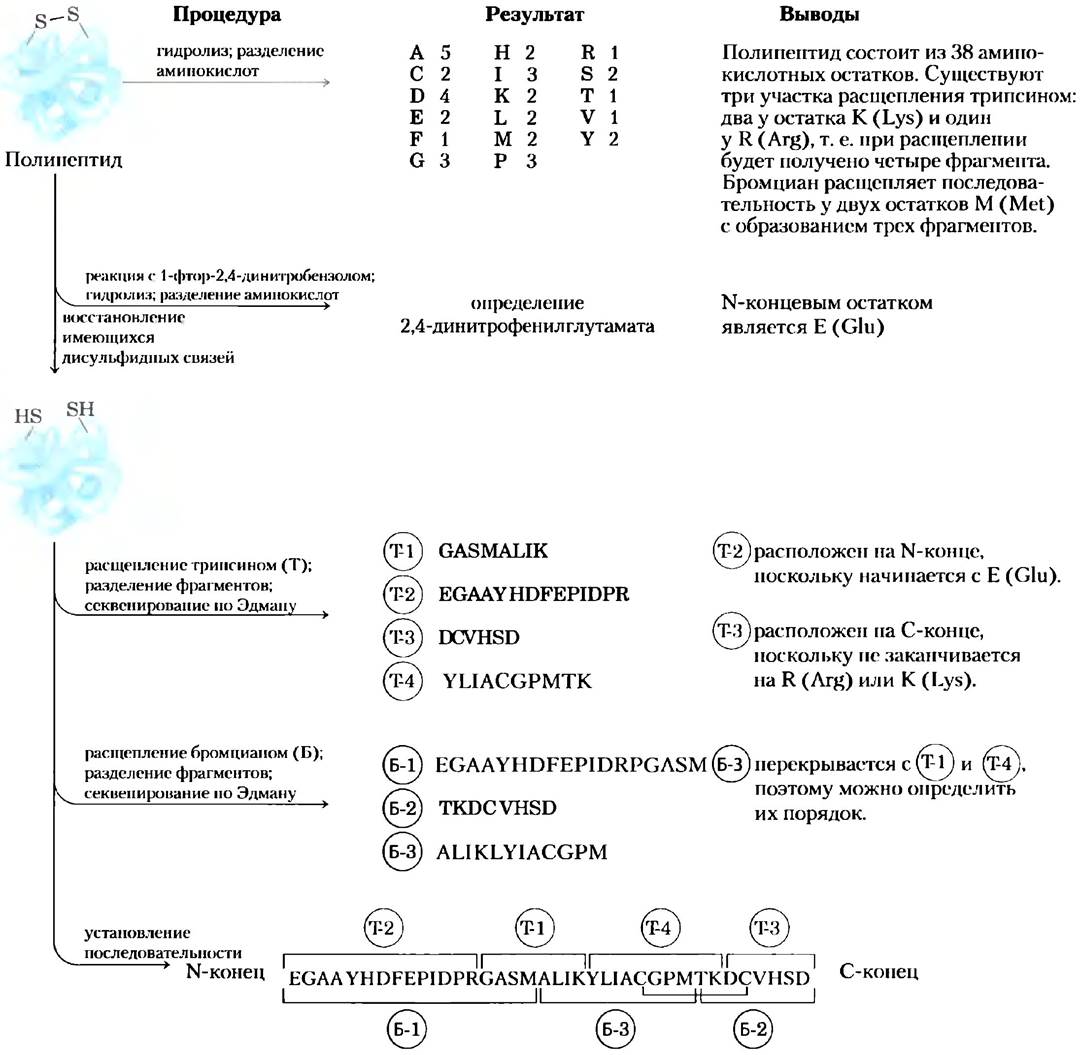
Секвенирование пептидов.Каждый фрагмент, полученный в результате растепления трипсином, секвенируют отдельно по методу Эдмана.
Определение порядка расположения фрагментов в исходном полипептиде. На данном этапе необходимо определить порядок расположения «трипсиновых фрагментов». Для этого образец исходного полипептида подвергают расщеплению еще одним ферментом или реагентом, который расщепляет пептидные связи между другими аминокислотными остатками, чем трипсин. Например, можно использовать бромциан, расщепляющий только те пептидные связи, карбонильная группа которых принадлежит остатку метионина. Полученные в результате расщепления фрагменты вновь очищают и секвенируют.
Затем необходимо исследовать аминокислотные последовательности всех имеющихся фрагментов с целью найти такие фрагменты, полученные в результате второго расщепления, которые перекрывали бы разрывы между фрагментами, полученными в первом эксперименте (рис. 3-27). Аминокислотная последовательность этих перекрывающихся пептидных участков позволяет установить порядок фрагментов, полученных в первом расщеплении. Если перед проведением первого расщепления была определена аминокислота, находящаяся на N-конце полипептида, то с помощью этой информации можно найти фрагмент, прилегающий к N-концу. Кроме того, сравнивая два набора фрагментов, можно найти возможные ошибки в определении аминокислотной последовательности. Иногда второго расщепления полипептида на фрагменты оказывается недостаточно, чтобы найти перекрывающиеся последовательности некоторых фрагментов. В этом случае применяют третий, а то и четвертый способ расщепления, что позволяет в итоге получить полный набор перекрывающихся последовательностей исходной цепи.
Локализация дисульфидных связей.Расположение дисульфидных связей в исходной молекуле определяют после завершения секвенирования последовательности. Для этого исходный полипептид вновь подвергают расщеплению, например, трипсином, на этот раз без предварительного разрушения дисульфидных связей. Образующиеся фрагменты разделяют методом электрофореза и сравнивают с набором фрагментов, полученных при первом расщеплении трипсином. Если между двумя фрагментами существует дисульфидная связь, то эти фрагменты отсутствуют в новом наборе фрагментов, зато там появляется более тяжелый фрагмент. Два отсутствующих пептида соответствуют участкам исходной полипептидной цепи, между которыми существует дисульфидная связь.
Аминокислотную последовательность можно вывести на основании других данных
Описанный выше метод не является единственным способом для установления аминокислотной последовательности. Новые масс-спектрометрические
методы позволяют определять последовательности коротких пептидов (20-30 аминокислотных остатков) всего за несколько минут (доп. 3-2). Кроме того, с развитием быстрых методов секвенирования ДНК (гл. 8), установлением генетического кода (гл. 27) и появлением методов идентификации генов (гл. 9) стало возможным определять аминокислотную последовательность полипептида на основании нуклеотидной последовательности кодирующего его гена (рис. 3-28). Методы определения последовательностей ДНК и белка дополняют друг друга. Для белка, ген которого доступен, секвенировапие ДНК может дать более быстрые и точные результаты, чем секвенирование самого белка. Аминокислотные последовательности большинства белков теперь определяют именно таким косвенным методом. Если ген не выделен, то необходимо проводить секвенирование белка, причем полученный результат дает больше информации, чем анализ последовательности ДНК, который, например, ничего не говорит о расположении в молекуле дисульфидных связей. Кроме того, знание аминокислотной последовательности даже небольшого участка полипептидной цепи может в значительной степени облегчить выделение соответствующего гена (гл. 9).
Рис. 3-28. Соответствие последовательностей ДНК и аминокислот. Каждая аминокислота кодируется специфической последовательностью трех нуклеотидов в цепочке ДНК. Подробно о генетическом коде см. гл. 27.
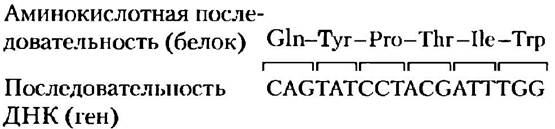
Практическая биохимия. Масс-спектрометрические методы изучения белков
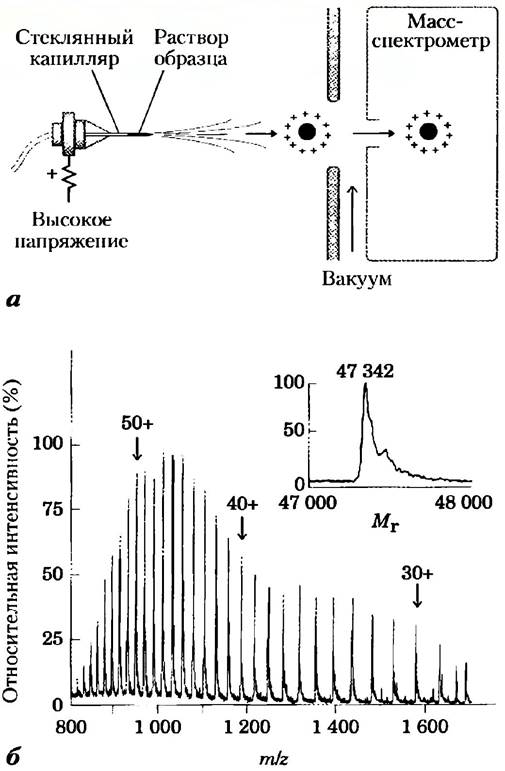
Масс-спектрометр является одним из важнейших приборов для химических исследований. Анализируемые молекулы (аналиты) сначала ионизируют в вакууме. Затем вновь образовавшиеся заряженные частицы вводят в электрическое и(или) магнитное иоле, причем их движение в поле зависит от отношения их массы к заряду (m/z). Эта измеряемая величина может быть использована для определения молекулярной массы (M) аналита с высокой точностью.
Масс-снектрометрия используется в качестве метода анализа уже долгие годы, но до относительно недавнего времени не было возможности использовать этот метод для изучения макромолекул, таких как белки и нуклеиновые кислоты. Дело в том, что измерение отношения т/г осуществляется в газовой фазе, а нагревание или другая процедура, необходимая для перевода вещества в газообразное состояние, обычно мечет за собой его разрушение. В 1988 г. были предложены два подхода, позволившие решить эту проблему. Водном методе белок помещают в светопоглощающую матрицу. Под действием пульсирующего лазерного излучения белки в матрице ионизируются и выходят из матрицы в вакуумную камеру. Этот метол, известный как лазерная десорбционно-ионизационная масс-спектрометрия (МАLDI), успешно используется для определения молекулярных масс широкого круга макромолекул. Во втором успешно применяющемся методе макромолекулы переходят в газовую фазу прямо из раствора. Раствор аналита пропускают через распылительную головку, находящуюся в сильном электрическом поле, в результате чего раствор превращается в мельчайшие заряженные капельки. Растворитель мгновенно испаряется, а заряженные макромолекулы в неповрежденном виде оказываются в газовой фазе. Данный метод носит название электроспрея (ЕSI- масс-спектрометрии). Протоны, присоединяющиеся к макромолекулам в процессе прохождения через распылительное устройство, сообщают им дополнительный заряд. Значение параметра т/г определяют в вакуумной камере.
Масс-спектрометрия позволяет получить большое количество информации, необходимой для исследований в области протеомики, энзимологии и химии белка в целом. Для проведения экспериментов требуются минимальные количества образцов: достаточное количество белка для анализа можно получить, например, методом двумерного электрофореза. Точное определение молекулярной массы белка - это один из ключевых моментов при сто идентификации. Если масса белка известна, то метод масс-спектрометрии позволяет детектировать все изменения, происходящие при связывании кофакторов, ионов металла, при ковалентных модификациях белка и т. д. На рис. 1 приведен пример определения молекулярной массы белка с помощью метода электроспрея.
Рис. 1. Метод электроспрея для определения молекулярной массы белка, а) Раствор белка распыляют в виде сильнозаряженных мельчайших капелек, пропуская через распыляющую головку, помещенную в сильное электрическое поле. Растворитель испаряется, а ионы аналита (в данном случае с присоединенными протонами) подаются в масс-спектрометр для определения отношения m/z. б) Полученный спектр представляет собой набор пиков, каждый из которых отличается от предыдущего (справа налево) увеличением массы и заряда на одну единицу. На вставке показан результат компьютерной обработки данного спектра.
При переходе в газовую фазу белок приобретает какое- то количество протонов (т. е. положительных зарядов) от молекул растворителя. В результате образуется спектр частиц с различным отношением массы к заряду. Каждый регистрируемый пик соответствует отдельному типу частиц, отличающемуся от соседнего на единицу заряда и единицу массы (на 1 протон). Молекулярную массу белка можно рассчитать по любым двум соседним пикам. Для одного пика:
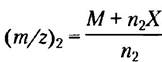
где М — молекулярная масса белка, n2 — число зарядов, X — молекулярная масса добавленной группы (в данном случае протона). Аналогично, для соседнего пика:
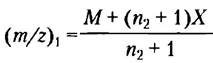
Теперь у нас есть два неизвестных (М и n2) и два уравнения, так что можем их решить сначала относительно n2, а затем относительно М:
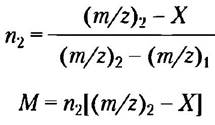
Такой способ вычислений на основании значений т/г для любых двух ников из спектра (рис. 1, б) обычно позволяет найти массу белка с ошибкой, не превышающей 0,01% (в данном случае анализировали белок аэролизин k с М = 47 342). Использование нескольких серий пиков, повторные вычисления и усреднение результатов могут еще более повысить точность определения молекулярной массы М. Компьютерные программы позволяют получить точный результат по значению m/z для одного пика (рис. 1, б, вставка).
Масс-снектрометрия является бесценным инструментом, с помощью которого можно быстро определить короткую аминокислотную последовательность неизвестного белка. Данный метод называется тандемной масс-спектрометрией. Сначала раствор исследуемого белка обрабатывают протеазой или химическим реагентом, чтобы получить смесь более коротких пептидов. Затем эту смесь подают в устройство, представляющее собой два последовательно расположенных масс-спектрометра
(рис. 2, а, вверху). В первом масс-спектрометре смесь ионизованных пептидов сортируется таким образом, чтобы только один тип фрагментов, полученных при расщеплении, мог достичь следующего отсека. Этот образец, каждая молекула которого имеет определенный заряд, проходит через вакуумную камеру между двумя масс-спектрометрами. В этой так называемой «камере соударений» пептид разрушается на еще более мелкие части под действием активных соударений с молекулами инертного газа (аргона или гелия), подаваемого в камеру. Процесс проводится в таких условиях, чтобы каждый отдельный пептидный фрагмент был разделен в среднем не более чем на две части. Большинство разрывов приходится на пептидные связи. В данной реакции молекулы воды нс участвуют, так как все происходит в вакуумной камере, поэтому среди продуктов реакции встречаются радикалы, например, карбонильные радикалы (рис. 2, а). Заряд, находившийся на исходном фрагменте, теперь сосредоточен на одной из его частей.
Рис. 2. Определение аминокислотной последовательности методом тандемной масс-спек- трометрии. а) Раствор белка после протеолитического расщепления вводят в масс-спектрометр (МС-1). Пептиды сортируют, отбирая для дальнейшего анализа лишь один тип фрагментов. Этот фрагмент далее подвергают фрагментации в камере, расположенной между двумя масс-спектрометрами. Во втором масс-спектрометре (МС-2) определяют значение m/z для каждого фрагмента. Многие образовавшиеся при фрагментации ионы получаются в результате разрыва пептидных связей. На рисунке они обозначены как ионы b-типа и y-типа в зависимости от того, остался заряд на С- или на N-конце фрагмента, б) Типичный спектр, полученный при обработке пептида, состоящего из 10 аминокислотных остатков. Помеченные пики соответствуют ионам y-типа. Самый высокий пик (соседний с пиком у5") соответствует иону с двойным зарядом и не относится к данной серии. Пики отличаются друг от друга на одну аминокислоту, стоящую рядом в пептидной цепи. В данном случае получена пептидная последовательность Phe-Pro-Gly-Gln-(Ile/Leu)-Asn-Ala-Asp-(Ile/Leu)-Arg. Заметьте, что существует неопределенность в идентификации лейцина и изолейцина, имеющих одинаковую молекулярную массу. В нашем примере серия пиков, относящихся к y-типу ионов, является преобладающей, что сильно облегчает расшифровку спектра. Это связано с тем, что на С-конце пептида расположен остаток Arg, на котором сосредоточена большая часть положительного заряда.
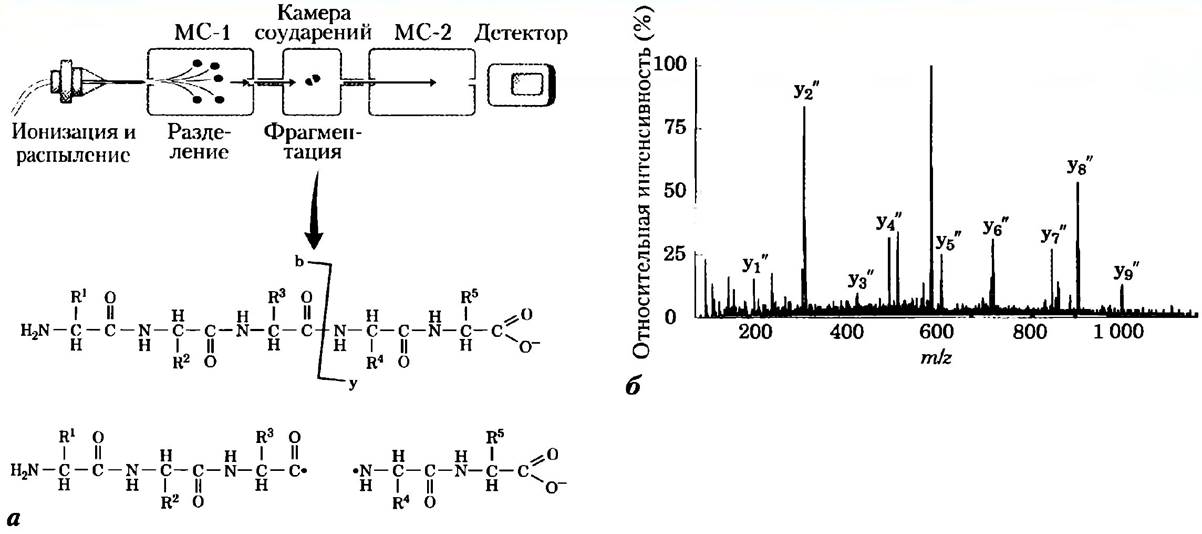
Во втором масс-спектрометре происходит измерение отношений т/г всех заряженных фрагментов (незаряженные частицы нс регистрируются). В результате получают одну или несколько серий пиков. Данная серия ников (рис. 2, б) отражает наличие всех заряженных фрагментов, которые образовались при разрыве одного и того же тина связи (но в разных частях полипептидной молекулы) с одной и той же стороны связи N или С). Соседние пики в одной серии различаются на одну аминокислоту. По разнице молекулярных масс соответствующих пиков можно определить, какая именно аминокислота стоит следующей в полипептидной цепи. Сложность возникает только с идентификацией лейцина и изолейцина, поскольку они имеют одинаковые молекулярные массы.
При разрушении пептида заряд может сохраняться как на N-концевом, так и на С-концевом фрагменте, кроме того, возможен распад молекулы не по пептидной связи, а в других местах, так что обычно в одном эксперименте получают несколько серий пиков. Два наиболее рельефных спектра обычно относятся к заряженным фрагментам, полученным при разрыве пептидных связей, причем набор С-концевых фрагментов очень легко отличить от набора N-концевых фрагментов. Дело в том, что
в процессе фрагментации в камере соударений в местах разрыва не образуется обычных карбоксильных или аминогрупп. Единственными полноценными α-амино- и α-карбоксильными группами являются группы, расположенные на самых концах фрагментов (рис. 2, а). В связи с этим, два набора фрагментов имеют небольшие различия в молекулярных массах. Определение аминокислотной последовательности по двум наборам фрагментов повышает точность результата.
Если известна нуклеотидная последовательность гена, то для однозначной» соотнесения гена с белком обычно достаточно прочесть лишь короткий фрагмент аминокислотной последовательности. Секвенирование с помощью масс-спектрометрии не может заменить секвенирования длинных фрагментов по методу Эдмана, но этот метод идеально подходит для исследований в области протеомики, направленных на каталогизацию сотен клеточных белков, которые можно разделить методом двумерного электрофореза.
Существующие сегодня методы анализа белков и нуклеиновых кислот дали возможность для развития новой дисциплины «биохимия целых клеток». Теперь можно изучать последовательности всей ДНК (генома) самых разных организмов — от вирусов и бактерий до многоклеточных организмов (табл. 1-2). Новые гены открывают тысячами, причем часть из них кодирует белки с пока неизвестными функциями. Для описания всего комплекса белков, кодируемых в геноме организма, ученые предложили использовать термин «протеом». Как будет обсуждаться далее в гл. 9, новые дисциплины протеомика и геномика — это взаимодополняющие разделы исследований в области клеточного метаболизма и метаболизма нуклеиновых кислот, направленные на создание все более полной картины биохимии клеток и целых организмов.
Небольшие пептиды и белки можно синтезировать химическим путем
Многие пептиды используются в фармакологии, поэтому их синтез имеет определенный коммерческий интерес. Есть три способа, с помощью которых можно получить пептид: 1) выделить из ткани, что часто затруднено его крайне низкой концентрацией; 2) получить генно-инженерным путем (гл. 9); 3) синтезировать химическим путем. Во многих случаях методы прямого химического синтеза являются предпочтительными. Кроме коммерческою приложения, химический синтез используется для создания специфических пептидных фрагментов, необходимых для изучения структуры и функций белков.
Традиционные методы синтеза органических соединений не годятся для получения пептидов и белков, имеющих в цепочке более 4 5 аминокислотных остатков, что связано с особой сложностью белковых структур. Одна из возникающих на этом пути проблем состоит в очистке продукта после его синтеза.
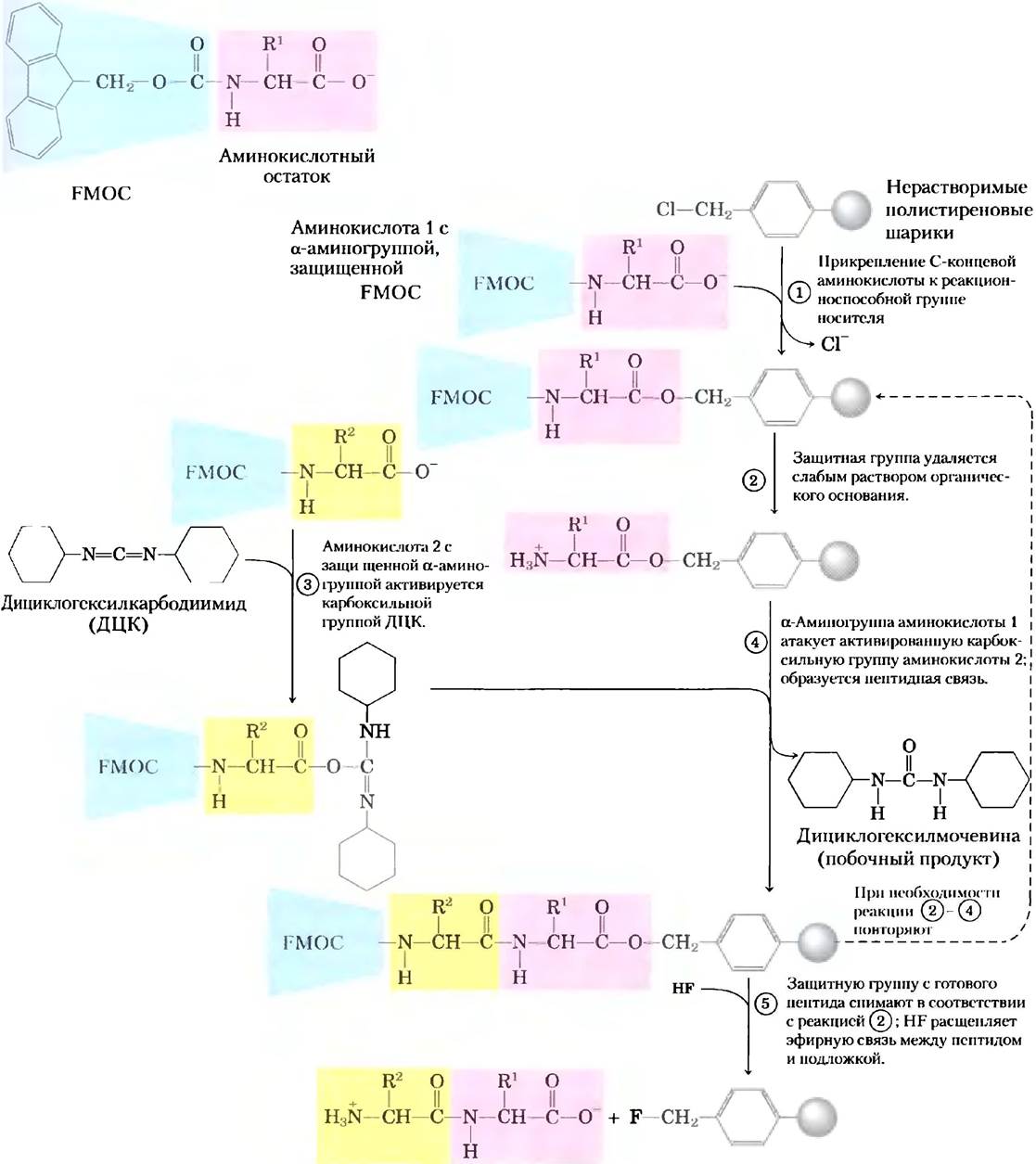
Большой вклад в развитие методов синтеза пептидов был сделан Робертом Брюсом Меррифил- дом в 1962 г. Он предложил синтезировать пептид, закрепив один его конец на твердой подложке. В качестве подложки можно использовать нерастворимый полимер (смолу), помещенный в колонку, похожую на те, что используются в хроматографии. К закрепленному на подложке пептиду с помощью стандартного набора реакций пришивают по одной аминокислоте, а затем повторяя цикл с другой аминокислотой (рис. 3-29). На каждой стадии цикла используют защитные химические группы, предотвращающие побочные реакции. Теперь технология химического пептидного синтеза автоматизирована. Аналогично реакциям секвенирования, данный процесс ограничивается эффективностью каждого отдельного реакционного цикла (это можно понять, рассчитав суммарную эффективность процесса, каждая стадия которого идет с выходом 96,0 или 99,8% (табл. 3-8)). Неполное проведение реакции на одной стадии приводит на следующей стадии к образованию примеси (более короткого пептида). Усовершенствование химических методов позволило с хорошим выходом синтезировать белки, состоящие из 100 аминокислот, всего за несколько дней. Очень близкий подход используется для синтеза нуклеиновых кислот (рис. 8-35). И все же методы лабораторного синтеза по-прежнему ничто по сравнению с биосинтезом в живых организмах! В бактериальной клетке такой же белок из 100 аминокислот синтезируется с высочайшей точностью приблизительно за пять секунд.
Таблица 3-8. Влияние выхода продукта на отдельных стадиях на общий выход пептидного синтеза
Число остатков в конечном полипептиде |
Общий выход готового продукта (%)в зависимости от 96,0% |
выхода на каждой стадии 99,8% |
11 |
66 |
98 |
21 |
44 |
96 |
31 |
29 |
94 |
51 |
13 |
90 |
100 |
1,8 |
82 |
Роберт Брюс Меррифилд, 1921-2006

Рис. 3-29. Химический синтез пептида на твердой подложке. Для образования каждой пептидной связи необходимо осуществление реакций 1-4. Защитная флуоренилметоксикарбонильная группа (FMОC, выделена синим цветом) предотвращает побочные реакции по α-аминогруппе аминокислотного остатка (выделены красным цветом). Химический синтез протекает в направлении от С-конца к N-концу пептидной цепи, т. е. в противоположном направлении по сравнению с синтезом in vivo (гл. 27).
Новые эффективные методы лигирования (сшивания) отдельных пептидных фрагментов позволяют собирать из синтетических пептидов более крупные белки. С помощью таких методов можно создавать новые белки, в которых химические группы, в том числе и не встречающиеся в природных белках, расположены в строго определенных местах. Новые белки позволяют изучать механизмы ферментативного катализа, создавать белки с новыми химическими свойствами и заданной структурой. Последнее окончательно убеждает нас в том, что есть связь между первичной структурой белка и его пространственной структурой в растворе.
Аминокислотная последовательность служит источником важной биохимической информации
Знание аминокислотной последовательности белка позволяет понять его трехмерную структуру, функции, локализацию в клетке и эволюцию. Важнейшую информацию можно получить, сравнивая аминокислотные последовательности данного белка и других известных белков. В Интернете есть доступ к базам данных, содержащим тысячи известных на сегодняшний день последовательностей. Сравнение новой последовательности с этим банком данных часто позволяет выявить как определенные особенности, так и общие закономерности.
До сих пор мы не знаем точно, каким образом аминокислотная последовательность определяет трехмерную структуру; мы также не в состоянии однозначно предсказать функции белка, исходя из его аминокислотной последовательности. Однако на основании сходства аминокислотной последовательности данного белка с другими белками его можно отнести к одному из известных семейств белков, отличающихся определенными функциональными и структурными особенностями. Члены семейства обычно имеют аминокислотные последовательности, совпадающие как минимум на 25%, а также обладают некоторыми общими структурными и функциональными характеристиками. Существуют, однако, семейства, члены которых имеют лишь несколько идентичных аминокислотных остатков, необходимых для реализации определенной функции. Кроме того, многие белки, выполняющие различные функции, имеют сходные надмолекулярные структуры (так называемые домены, см. гл. 4). Эти домены часто образуют специфические структуры, обладающие неожиданно высокой устойчивостью или приспособленные к существованию в определенном окружении. Кроме того, на основании структурного и функционального сходства между семействами белков можно сделать определенные заключения относительно их эволюционного развития.
Некоторые аминокислотные последовательности служат сигналами, определяющими их клеточную локализацию, необходимости химической модификации и указывают на время жизни белков. Специальные сигнальные последовательности, обычно расположенные на N-конце белка, используются для маркировки белков, транспортируемых за пределы клетки; существуют также белки, предназначенные для локализации в ядре, на клеточной поверхности или в других отделах клетки. Определенные последовательности служат участками связывания простетических групп, таких как остатки сахаров в гликопротеинах и липиды в липопротеинах. Некоторые из этих сигнальных последовательностей хорошо изучены и легко распознаются в аминокислотной последовательности нового белка (гл. 27).
Консенсусные последовательности и Sequence logos
Консенсусные последовательности можно изобразить несколькими различными способами. Для иллюстрации двух вариантов представления данных мы используем два примера консенсусных последовательностей, изображенных на рис. 1: а) АТР-связывающая структура, называемая Р-петлей (см. доп. 12-2); б) Са2+-связывающая структура, называемая EF-рукой (см. рис. 12-11). Приведенные здесь правила — адаптированный вариант правил, используемых веб-сайтом PROSITE (expasy.org/prositc); в этой программе для обозначения аминокислотных остатков применяется однобуквенный код.
Рис. 1. Представление двух консенсусных последовательностей. а) Р-петля — АТР-связывающая структура; б) ЕF-рука — Са2+-связывающая структура.

В одном способе представления консенсусных последовательностей (верхняя часть рисунков а и б) каждая позиция отделена от соседней позиции черточкой. Позиция, в которой может находиться любая аминокислота, обозначается через х. В случае неопределенности в квадратных скобках перечисляются все возможные аминокислоты. Например, в случае a [AG] означает Ala или Gly. Если в какой- либо позиции могут находиться практически любые аминокислоты за некоторым исключением, то на месте этой позиции в фигурных скобках перечисляют именно те аминокислоты, которые нс могут здесь стоять. Например, на рис. 1,6 {W} означает, что здесь не может находиться аминокислота Тrр. Повторение какого-либо элемента последовательности обозначают с помощью числа или нескольких чисел, взятых в скобки и стоящих после этого элемента. Так, в примере (а) х (4) означает х-х-х-х, а (2,4) означает х-х, х-х-х или х-х-х-х. Если рассматриваемый участок является N-концом или С-концом последовательности, то запись соответственно начинают или заканчивают им (это не так в данных примерах). В конце фрагмента ставится точка. Применяя эти правила к консенсусной последовательности (а), получаем, что в первой позиции может стоять А или G. В следующих четырех позициях могут располагаться любые аминокислоты, далее обязательно идут G и К, а последнюю позицию занимает S или Т.
Система записи Sequence logos обеспечивает возможность более информативного графического представления выравнивания множества аминокислотных (или нуклеотидных) последовательностей. В каждой позиции указан определенный набор символов, соответствующий тем аминокислотам (или нуклеотидам), которые могут здесь располагаться.
Общая высота символа в позиции (в битах) отражает степень консервативности этой позиции, а высота каждого отдельного символа внутри множества указывает на относительную частоту встречаемости именно этой аминокислоты (нуклеотида). В аминокислотных последовательностях цветом обозначены характеристики аминокислот: полярные аминокислоты (G, S, Т, Y, С, Q, N) зеленые, основные (К, R, Н) синие, кислые (D, Е) красные, а гидрофобные (А, V, L, I, Р, W, F, М) черные. Классификация аминокислот в данной схеме несколько отличается от той, что представлена в табл. 3-1 и на рис. 3-5. Аминокислоты с ароматической структурой боковой цепи относят как к неполярным (F, W), так и к полярным (Y). Глицин, который всегда трудно поддается классификации, принято считать полярным. Если в одной позиции могут находиться одна или небольшое число аминокислот, редко бывает так, что они встречаются в этой позиции с равной вероятностью. Одна или несколько аминокислот обычно преобладают. Данный способ представления делает это преобладание наиболее выраженным, а консервативную последовательность в белке — более очевидной. Однако при этом скрываются некоторые аминокислоты, которые могли бы находиться в определенной позиции, например, Суs, который иногда встречается в позиции 8 в мотиве ЕF-руки (рис. 1, б).
Ключевые договоренности.
Значительная доля функциональной информации, содержащейся в последовательности белка, заключена в консенсусных последовательностях. Этим термином обозначают соответствующие последовательности не только в белке, но также в ДНК и РНК. При сравнении наборов родственных аминокислотных или нуклеотидных последовательностей выявляют позиции, в которых чаще всего стоят одни и те же аминокислоты или нуклеотиды; это и есть консенсусные последовательности. Участки последовательности, отличающиеся наибольшим сходством в ряде организмов, часто соответствуют эволюционно консервативным функциональным доменам. Для анализа и поиска консенсусных последовательностей создан целый набор математических приемов и программ, доступных через Интернет. В доп. 3-3 представлены общие принципы изображения консенсусных последовательностей. ■
Белковые последовательности проливают свет на развитие жизни на Земле
Простая последовательность букв в аминокислотной последовательности не дает полного представления о том богатстве информации, которое на самом деле заключено в этой последовательности. По мере роста числа известных белковых последовательностей появляются все более мощные методы, позволяющие извлечь из них самую разную информацию. Анализ информации, хранящейся в постоянно расширяющихся биологических базах данных, содержащих последовательности генов и белков, а также структуры макромолекул, привел к становлению биоинформатики как новой области знаний. Один из результатов развития этой дисциплины заключается в появлении серий компьютерных программ, многие из которых доступны через Интернет и их могут использовать ученые, студенты и даже непрофессионалы. Функции каждого белка связаны с его трехмерной структурой, которая, в свою очередь, во многом определена первичной последовательностью. Таким образом, биохимическая информация, отраженная в аминокислотной последовательности, в принципе ограничена нашими собственными знаниями о структурных и функциональных принципах организации белков. Постоянно развивающийся аппарат биоинформатики позволяет идентифицировать функциональные сегменты в новых белках и установить, как их последовательность, так и их структурную связь с уже известными белками. Кроме того, изучение белковых последовательностей с эволюционной точки зрения позволяет нам понять, как возникали белки и, в конечном итоге, как развивалась жизнь на нашей планете.
Историю молекулярной эволюции обычно связывают с работами Лайнуса Полинга и Эмиля Цукеркандла, которые в середине 1960-х гг. стали активно использовать нуклеотидные и аминокислотные последовательности для изучения эволюционного процесса. Основа этой науки обманчиво проста. Если два организма родственны друг другу, то последовательности их генов и белков должны быть похожи. По мере эволюционного расхождения организмов эти последовательности начинают сильно различаться. Перспективы развития такого подхода стали проясняться в 1970-х гг., когда Карл Вёзе использовал последовательности рибосомных РНК для выделения архей в отдельную группу, не принадлежащую к бактериям или эукариотам (рис. 1-4). Белковые последовательности часто предоставляют возможность для уточнения имеющейся информации. По мере реализации проектов по изучению геномов самых разных организмов от бактерии до человека число доступных последовательностей растет с огромной скоростью. Эту информацию можно использовать, чтобы проследить ход эволюции. Проблема заключается в расшифровке генетических иероглифов.
Эволюция не следует простым линейным путем. Сложности возникают при каждой попытке извлечь связанную с эволюцией информацию, заключенную в белковых последовательностях. В каждом конкретном белке на протяжении всего хода эволюции неизменными оставались аминокислотные остатки, имеющие наибольшее
значение для его функционирования. Остатки, не играющие такой важной роли в активности белка, могли со временем меняться, т. е. одна аминокислота могла заменить другую, и именно эти изменившиеся остатки могут сказать что-то о ходе эволюционного процесса. Но аминокислотные замены не всегда случайны. В некоторых участках аминокислотной последовательности возможны только строго определенные замены, что связано с необходимостью сохранения функции белка. Аминокислотный состав некоторых белков изменялся сильнее, чем других. По этим и другим причинам скорость, с которой белки эволюционируют, разная.
Еще один фактор, мешающий проследить ход эволюции, — перенос генов или групп генов от одного организма в другой, называемый горизонтальным (латеральным)переносом генов. Перенесенные гены могут быть довольно похожими на те, что были в исходном организме, в то время как большинство остальных генов в этих двух организмах имеют лишь весьма отдаленное сходство. Результатом горизонтального переноса генов является наблюдающееся сегодня быстрое распространение устойчивости к антибиотикам в популяции бактерий. Белки, синтезируемые на основе этих перенесенных генов, будут неудачными кандидатами для изучения эволюции бактерий, поскольку история их эволюции в новом «хозяйском» организме очень коротка.
Предметом исследования в молекулярной эволюции обычно служат семейства близкородственных белков. Для анализа отбирают семейства белков, играющих важную роль в клеточном метаболизме, поскольку они обязательно должны были присутствовать и в клетках-предшественниках, а это сильно снижает вероятность их появления в клетках в результате горизонтального переноса генов. Например, белок еЕF-1α (фактор элонгации 1α) участвует в синтезе белка у всех эукариот. Похожий белок ЕF-Тu обнаружен в клетках бактерий. Сходство последовательностей и выполняемых функций говорит о том, что еЕF-α1 и ЕF-Тu являются членами семейства белков, произошедшего от общего предшественника. Белки, входящие в состав семейства, называют гомологичными белками, или гомологами. Если два члена семейства, т. е. два гомолога, присутствуют в организмах одного вида, их называют паралогами. Гомологичные белки из организмов разных видов называют ортологами. Для изучения процесса эволюции в первую очередь необходимо идентифицировать подходящие семейства гомологичных белков, а затем уже на их основе восстановить ход эволюции.
Идентификация гомологов осуществляется с помощью мощных компьютерных программ, которые позволяют напрямую сравнивать две или несколько белковых последовательностей, а также осуществлять поиск близких последовательностей в базах данных. Процесс электронного поиска можно представить себе, как скольжение одной последовательности по другой до того момента, пока не будет найден участок достаточного сходства. В данном процессе выравнивания последовательностей каждая позиция, в которой аминокислоты из двух аминокислотных последовательностей совпадают, получает определенный вес, различающийся в зависимости от использованной компьютерной программы; этот вес служит показателем качества выравнивания. На данном пути возникают определенные сложности. Иногда сравниваемые белки достаточно хорошо совпадают, скажем, на двух участках последовательностей, разделенных между собой менее схожими участками разной длины. В результате одновременное выравнивание двух совпадающих участков невозможно. Для решения этой проблемы компьютерная программа вводит в одну из последовательностей пробел таким образом, чтобы два схожих участка могли накладываться друг на друга одновременно (рис. 3-30). Очевидно, что с использованием достаточного количества пробелов практически любые две последовательности можно некоторым образом выровнять. Чтобы избежать неинформативного выравнивания, компьютерная программа за каждый пробел вычитает определенный штраф, гем самым снижая вес выравнивания. Далее программа отбирает оптимальное выравнивание, при котором число идентичных аминокислотных остатков максимально, а число пробелов минимально.
Рис. 3-30. Выравнивание белковых последовательностей с использованием пробелов. Здесь представлены короткие участки белка Hsp70 (широко распространенного класса белков-шаперонов) из двух хорошо изученных бактерий - Е. coli и Bacillus subtilis. Введение пробела в аминокислотную последовательность В. subtilis позволяет лучше осуществить выравнивание с каждой стороны от пробела. Желтым цветом выделены идентичные аминокислотные остатки.
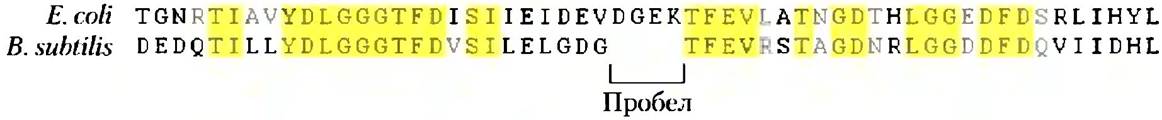
Расположение идентичных аминокислот часто не позволяет определить степень родства белков в эволюционном плане. Более полезно в этом смысле исследование химических свойств аминокислотных замен. Аминокислотные замены в семействе белков часто бывают консервативными; это означает, что один аминокислотный остаток заменен другим, но с близкими химическими свойствами. Например, в одном белке семейства в определенной позиции находится Glu, а в другом — Asp; заметьте, что оба остатка несут отрицательный заряд. Такая консервативная замена из логических соображений позволит осуществить более надежное выравнивание, чем неконсервативная замена того же Asp, например, на гидрофобный остаток Phe.
В большинстве случаев для поиска гомологов и выяснения эволюционного родства белковые последовательности (как полученные в результате прямого аминокислотного секвенирования, так и выведенные на основании последовательности ДНК) более предпочтительны, чем некодирующие нуклеотидные последовательности (те, которые не кодируют последовательности белка или функциональной РНК). В случае нуклеиновых кислот, построенных всего из четырех различных остатков, случайное выравнивание негомологичных последовательностей обычно приводит к совпадению как минимум 25% позиций. Введение нескольких пробелов часто может повысить эту значение до 40% и более; в результате вероятность выравнивания неродственных последовательностей очень высока. Наличие 20 различных аминокислот в белке значительно снижает возможность неинформативного выравнивания подобного рода.
Все программы выравнивания последовательностей снабжены методами тестирования надежности выравнивания. В одном из тестов аминокислотную последовательность одного из сравниваемых белков перемешивают для получения случайной последовательности, а затем запускают программу выравнивания для сравнения се с исходной последовательностью. Новое выравнивание вновь характеризуют весами, а процессы перемешивания и выравнивания можно повторять много раз. Первое выравнивание, осуществленное до перемешивания, должно иметь гораздо больший вес, чем выравнивания случайных последовательностей. В таком случае существует уверенность, что выровненные последовательности действительно принадлежат двум гомологам. Заметьте, что отсутствие значительного веса выравнивания не обязательно означает отсутствие эволюционной связи двух белков. Как мы увидим в г л. 4, иногда при изучении трехмерных структур становится очевидным эволюционное родство, хотя гомология последовательностей не была обнаружена.
Для изучения эволюционного родства стараются использовать семейства белков со сходными функциями из максимально возможного диапазона организмов. Полученная информация может быть использована для слежения за ходом эволюции этих организмов. На основании анализа расхождений в выбранных семействах белков исследователь может разделить организмы на классы в соответствии с их эволюционными связями. Эти данные должны соответствовать результатам классических исследований по физиологии и биохимии организмов.
Некоторые участки белковой последовательности могут встречаться у организмов, относящихся к одной таксономической группе, но не к другим группам. Эти участки можно использовать в качестве маркерных последовательностей для тех групп, в которых они были обнаружены. Примером такой маркерной последовательности является вставка из 12 аминокислот в N-концевой области белков еЕF-α1/ЕF-Тu у всех архей и эукариот, но не у бактерий (рис. 3-31). Маркерные последовательности являются одним из тех ключей, которые помогают установить эволюционную связь между эукариотами и археями. Другие характерные последовательности позволяют установить эволюционные связи между группами организмов на многих таксономических уровнях.
Рис. 3-31. Маркерная последовательность семейства белков еЕF-α1/ЕF-Тu. Маркерная последовательность (обведена рамкой) представляет собой вставку из 12 аминокислотных остатков, расположенную в N-концевой области. Совпадающие во всех последовательностях основания выделены желтым цветом. Маркерная последовательность существует как у архей, так и у эукариот, хотя в двух группах организмов эти вставки довольно сильно различаются. Изменения внутри маркерной последовательности отражают значительное эволюционное расхождение, которое произошло на этом участке после того, как он впервые появился у общего предшественника обеих групп.

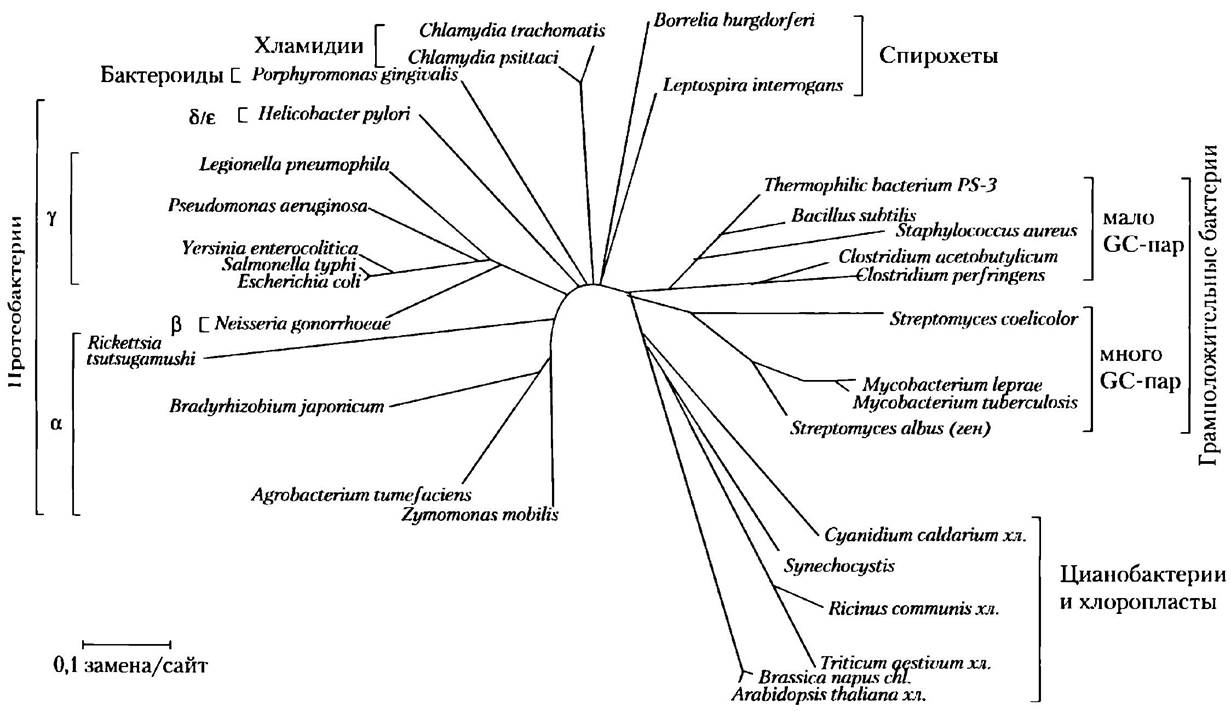
Анализируя полные аминокислотные последовательности белков, можно построить более совершенные эволюционные древа со многими видами в каждой таксономической группе. На рис. 3-32 показано такое древо для бактерий, построенное на основании расхождений в последовательности белка GrоEL (белок, присутствующий во всех бактериях и необходимый для нормального фолдинга). Древо можно дополнительно усовершенствовать, используя анализ последовательностей многих белков, а также привлекая данные об уникальных биохимических и физиологических свойствах организмов каждого вида. Существует множество способов для построения эволюционного древа, каждый из которых имеет как преимущества, так и недостатки, а также множество способов представления эволюционных отношений. Окончания ветвей на древе, изображенном на рис. 3-32, соответствуют существующим в наши дни видам, названия которых указаны. Точки, в которых сходятся две линии, соответствуют их вымершему предку. В большинстве способов изображения деревьев, в том числе и на рис. 3-32, длина линий между точками пропорциональна числу аминокислотных замен, отличающих один вид от другого. Таким образом, если мы рассмотрим точки, соответствующие двум существующим видам и их общему предшественнику, то длина каждой линии, соединяющей окончание ветви с местом ее ответвления, соответствует числу аминокислотных замен, отличающих каждый существующий вид от предшественника. Сумма длин линий, исходящих из точки ветвления к концам ветвей, отражает число аминокислотных замен, на которое эти существующие ныне организмы отличаются между собой. Чтобы определить промежутки времени, необходимые для расхождения видов, древо следует откалибровать в соответствии с данными анализа ископаемых остатков и с другой имеющейся информацией.
Рис. 3-32. Эволюционное древо, построенное на основании сравнения аминокислотных последовательностей. Здесь представлено эволюционное древо бактерий, основанное на расхождении аминокислотных последовательностей семейства белков GrоЕL. Кроме того, на древе указано расположение хлоропластов (хл.) некоторых других видов организмов.
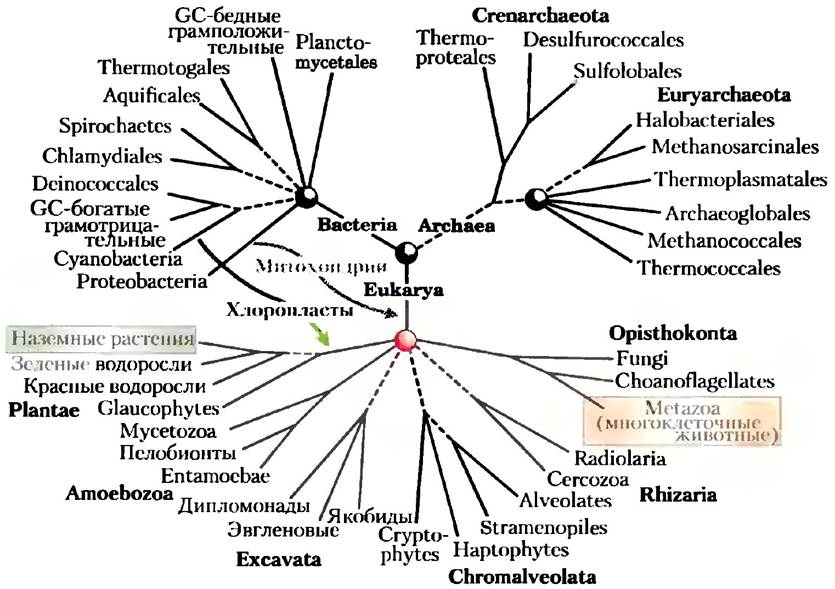
По мере увеличения объема информации в банках данных становится возможным построение эволюционных деревьев на основании последовательностей все большего числа различных белков. Благодаря все более усложняющимся методам анализа продолжает возрастать количество генетической информации, так что структура этих деревьев может быть дополнительно уточнена. Все подобные работы продвигают нас к цели создания подробного дерева жизни, описывающего эволюцию и взаимоотношения всех организмов на Земле. Развитие данной области науки продолжается (рис. 3-33); поставленные в ней вопросы важны для осознания человеком самого себя и окружающего мира. Молекулярная эволюция обещает быть одной из наиболее живых отраслей науки двадцать первого века.
Рис. 3-33. Консенсусное древо жизни. Показанное здесь древо основано на анализе многих белковых последовательностей и дополнительных особенностей генома. Ветви, обозначенные пунктиром, еще исследуются. Такое древо представляет только часть доступной информации, и только часть проблем нашла в нем свое решение. Каждая ныне живущая группа имеет свою собственную сложную историю эволюции.
Краткое содержание раздела 3.4 Структура белка: первичная структура
■ Различные функции белков определяются их аминокислотным составом и аминокислотной последовательностью. Некоторые небольшие изменения аминокислотной последовательности белка могут не сопровождаться изменениями его функций.
■ Определение аминокислотной последовательности пептида происходит в несколько этапов: 1) разделение полипептида на более мелкие фрагменты с помощью реагентов, расщепляющих специфические пептидные связи; 2) определение аминокислотной последовательности каждого фрагмента по методу Эдмана; 3) определение расположения фрагментов в исходной полипептидной цепи с помощью создания другого набора перекрывающихся фрагментов. Аминокислотную последовательность, кроме того, можно вывести на основании известной нуклеотидной последовательности соответствующего гена.
■ Короткие белки и пептиды (до 100 остатков) можно синтезировать химическим путем. На каждом этапе синтеза происходит присоединение одной аминокислоты, причем вся пептидная цепочка постоянно удерживается на твердом носителе.
■ Аминокислотная последовательность белка является важным источником информации не только о структуре и функции белка, но и об эволюции жизни на нашей планете. Разработаны сложные методы, позволяющие проследить ход эволюции на основании анализа изменений аминокислотных последовательностей гомологичных белков.