ОСНОВЫ БИОХИМИИ ЛЕНИНДЖЕРА - ТОМ 1. ОСНОВЫ БИОХИМИИ СТРОЕНИЕ И КАТАЛИЗ - 2011
ЧАСТЬ I. СТРОЕНИЕ И КАТАЛИЗ
6. ФЕРМЕНТЫ
6.2. Как работают ферменты
Ферментативный катализ реакций играет важнейшую роль в живых системах. При стандартных физиологических условиях некатализируемые реакции идут крайне медленно, поскольку большинство биологических молекул достаточно устойчивы при нейтральных значениях pH и температуре, при которой обычно существуют живые клетки. Более того, во многих биохимических реакциях происходят события, являющиеся довольно неблагоприятными или маловероятными в клетке, например, временное образование неустойчивых промежуточных продуктов или столкновение двух, или большего числа молекул в строго определенной ориентации. Реакции, необходимые для переваривания пищи, передачи нервного сигнала или мышечного сокращения, просто-напросто не могли бы протекать с ощутимыми скоростями, если бы не было катализаторов.
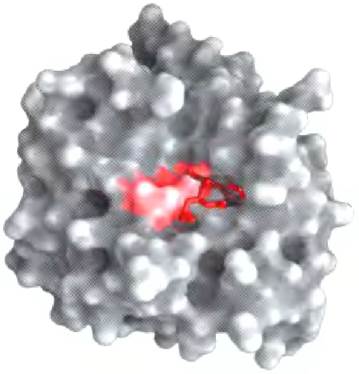
Фермент преодолевает эти препятствия, создавая специфические условия, при которых данная реакция может идти быстрее. Отличительной особенностью ферментативной реакции является то, что она происходит внутри кармана на поверхности фермента, называемого активным центром (рис. 6-1). Молекула, связывающаяся в активном центре и подвергающаяся воздействию фермента, называется субстратом. Поверхность активного центра образована аминокислотными остатками, способными связывать субстрат и катализировать его химическое превращение. Часто в результате связывания субстрат скрывается в активном центре фермента и полностью отделяется от остального раствора. Образование комплекса фермента с субстратом, существование которого впервые предсказал Шарль-Адольф Вюрц в 1880 г., — ключевая стадия ферментативного катализа. Этот комплекс, кроме того, является отправной точкой в математических расчетах, описывающих кинетические закономерности ферментативных реакций, а также в теоретических представлениях о механизмах ферментативного катализа.
Рис. 6-1. Связывание субстрата в активном центре фермента. Здесь представлена модель химотрипсина (РDВ ID 7GСН) со связанным субстратом (красного цвета). Некоторые ключевые аминокислоты активного центра изображены в виде красного пятна на поверхности фермента.
Ферменты влияют на скорость реакции, не сдвигая равновесия
Простая ферментативная реакция в общем виде записывается следующим образом:
Е + S ⇄ ЕS ⇄ ЕР ⇄ Е + Р (6-1)
где Е, S и Р — фермент, субстрат и продукт реакции соответственно; ЕS и ЕР — промежуточные комплексы фермента с субстратом и фермента с продуктом.
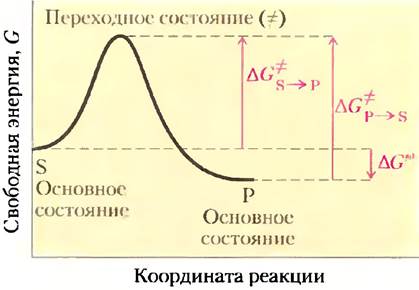
Чтобы говорить дальше о катализе, нам следует четко разобраться в понятиях «равновесие реакции» и «скорость реакции». Функция катализатора состоит в увеличении скорости реакции, но он не может влиять на равновесие ускоряемой им реакции. Любую реакцию, например, S ⇄ Р, можно описать с помощью энергетической диаграммы —графика изменения энергии вдоль координаты реакции (рис. 6-2). Как обсуждалось в гл. 1, биологические системы характеризуются величиной свободной энергии (G). Итак, ход реакции можно изобразить с помощью зависимости свободной энергии системы от координаты реакции. Исходная точка прямой и обратной реакции называется основным состоянием и определяет вклад молекулы S или Р в свободную энергию системы в конкретных условиях.
Рис. 6-2. Энергетическая диаграмма химической реакции. График описывает изменение свободной энергии системы в течение реакции S —> Р. В диаграммах подобного рода по оси ординат откладывают энергию системы, а по оси абсцисс — координату реакции, отражающую последовательные изменения в системе по мере превращения S в Р (например, образование и разрыв связей). На графике показаны значения энергии активации (∆G°) для реакций S —> Р и Р —> S. ∆G° — стандартная свободная энергия реакции S —> Р.
Ключевые договоренности.
Для описания изменений свободной энергии необходимо задать стандартные условия: температура 298 К, парциальное давление любого газа 1 атм (101,3 кПа), концентрации всех растворенных веществ 1 М; изменения свободной энергии для данной реакционной системы следует записать как изменение стандартной свободной энергии (∆G°). Поскольку биохимические процессы обычно протекают при концентрации Н+ значительно ниже 1 М, в биохимии используют изменение стандартнойбиохимической свободной энергии (∆G′°), что соответствует ∆G° при pH 7,0. Далее здесь мы будем пользоваться именно этим параметром. Более полное определение ∆G°приводится в гл. 13. ■
Положение равновесия между S и Р зависит от разности значений свободной энергии основных состояний этих веществ. В примере, изображенном на рис. 6-2, свободная энергия вещества Р в основном состоянии ниже, чем вещества S, поэтому ∆G′° данной реакции имеет отрицательное значение, и равновесие сдвинуто в сторону образования Р (реакция протекает слева направо). Положение равновесия реакции не зависит от наличия катализатора.
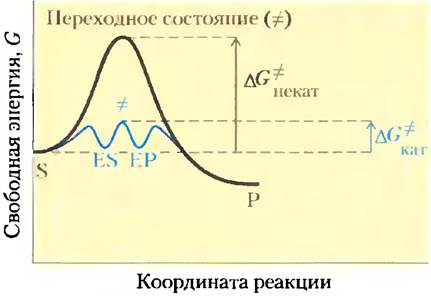
Благоприятные термодинамические параметры, однако, не означают, что превращение S в Р протекает с измеримой скоростью. Скорость реакции зависит совсем от других величин. Вспомним, что на пути перехода от S к Р существует энергетический барьер, поскольку для протекания реакции в любом направлении необходимо затратить энергию на правильную ориентацию реакционных групп, образование неустойчивого соединения, перегруппировку связей в молекулах и другие преобразования. На рис. 6-2 и 6-3 это отражено в виде пика свободной энергии. Чтобы реакция могла протекать, молекулы должны обладать достаточной энергией для преодоления этого барьера. На вершине энергетического пика существует точка, в которой вероятность протекания реакции в обоих направлениях одинакова. Эта точка соответствует переходному состоянию.Переходному состоянию не отвечает определенная стабильная химическая структура; его не следует путать с промежуточными продуктами реакции (ЕS и ЕР). Это лишь определенный короткий отрезок времени, на протяжении которого происходит разрушение связей, образование связей и локализация заряда, причем в этот момент вероятность образования исходного вещества и продукта реакции одинакова. Разность между значениями энергии в основном и переходном состояниях называется энергией активации (∆G°). Скорость реакции связана с энергией активации: чем выше энергия активации, тем медленнее протекает реакция. Увеличить скорость реакции можно, подняв температуру, поскольку при этом возрастает количество молекул, обладающих достаточной энергией для преодоления энергетического барьера. Другой способ ускорить реакцию состоит в добавлении катализатора (рис. 6-3). Катализатор ускоряет реакцию путем снижения энергии активации.
Рис. 6-3. Энергетическая диаграмма реакции в присутствии катализатора и без него. В реакции S —> Р промежуточные соединения ЕS и ЕР занимают энергетические минимумы, образующиеся в процессе катализируемой ферментом реакции. Энергии активации некатализируемой и катализируемой реакций обозначены соответственно ∆G*некат и ∆G*кат. Видно, что в случае ферментативной реакции энергия активации ниже.
Ферменты, как и все остальные катализаторы, не сдвигают равновесие реакции. Знак обратимости в уравнении 6-1 подчеркивает этот принцип: фермент, катализирующий реакцию S —> Р, также катализирует и реакцию Р —> S. Роль фермента заключается в ускорении прямой и обратной реакций. Сам фермент при этом не расходуется, и равновесие реакции не сдвигается. Но в присутствии соответствующего фермента реакция достигает равновесия гораздо быстрее, поскольку скорость реакции возрастает.
Этот общий принцип можно проиллюстрировать на примере взаимодействия сахарозы с кислородом с образованием углекислого газа и воды:
С12Н22O11 + 12O2 ⇄ 12СO2 + 11Н2O
Это превращение, протекающее в несколько стадий, характеризуется большим по абсолютной величине отрицательным значением ∆G°′, поэтому в состоянии равновесия количество сахарозы в реакционной смеси незначительно. Тем не менее сахароза является устойчивым соединением, поскольку энергетический барьер, который нужно преодолеть для взаимодействия сахарозы с кислородом, достаточно высокий. В присутствии кислорода сахароза хранится бесконечно долго, не подвергаясь заметным превращениям. Однако в клетках сахароза быстро распадается на СO2 и Н2O в результате серии ферментативных реакций. Участвующие в этих реакциях ферменты не только ускоряют их, но также организуют и контролируют таким образом, что большая часть высвобождаемой в результате реакций энергии извлекается в другой химической форме и может быть использована в клетке для других целей. Расщепление сахарозы и других сахаров является для клетки основным источником энергии; с участием ферментов соответствующие реакции протекают с такой скоростью, чтобы удовлетворить потребности клетки в энергии.
Практически все реакции имеют несколько промежуточных стадий, на которых происходит образование и распад весьма недолговечных химических соединений, называемых промежуточными соединениями или интермедиатами* реакции. Интермедиаты образуются в ходе реакции и имеют ограниченное время жизни (более длительное, чем время колебания молекулы, равное -10-13 с). Если реакция S = Р катализируется ферментом, то интермедиатами данного процесса могут быть комплексы ЕS и ЕР, несмотря на то что S и Р являются устойчивыми химическими молекулами (см. уравнение 6-1). Комплексам ЕS и ЕР отвечают минимумы на энергетической диаграмме (рис. 6-3). Кроме того, в ферментативной реакции могут возникать и менее устойчивые интермедиаты. Взаимное превращение двух последовательных интермедиатов составляет одну стадию реакции. Если реакция протекает в несколько стадий, ее общая скорость определяется скоростью той стадии (или тех стадий), которая имеет максимальную энергию активации; эту стадию называют лимитирующей стадией. В простейшем случае лимитирующая стадия соответствует максимуму энергии на диаграмме взаимопревращений S и Р. Какая стадия будет лимитирующей, зависит от условий проведения реакции. Для многих ферментов сразу несколько стадий могут иметь одинаковые энергии активации, что означает, что все они являются лимитирующими.
* В данной главе понятия «стадия» и «интермедиат» относятся к химическим соединениям, образующимся в процессе с участием одного фермента. При обсуждении метаболических путей, в которых задействовано множество ферментов (см. часть II), эти термины используют несколько иначе. Одну ферментативную реакцию часто называют стадией метаболического пути, а продукт ферментативной реакции, являющийся субстратом для следующей реакции, называют интермедиатом.
Энергия активации представляет собой энергетический барьер химической реакции. Наличие таких барьеров играет чрезвычайно важную роль в живых организмах. Скорость, с которой молекула претерпевает определенные изменения, снижается при повышении энергии активации. Без подобных энергетических барьеров сложные макромолекулы самопроизвольно превращались бы в более простые молекулярные формы, и существование сложных высокоорганизованных структур, а также протекание метаболических процессов в клетке не представлялось бы возможным. В ходе эволюции возникли ферменты, способные избирательно понижать энергии активации тех реакций, которые необходимы клетке.
Скорость реакции и равновесие связаны с определенными термодинамическими параметрами
Равновесие реакции неразрывно связано со стандартной свободной энергией реакции (∆G'°), а скорость реакции — с энергией активации (∆G'°). Для дальнейшего изучения принципов ферментативного катализа необходимо несколько подробнее остановиться на этих термодинамических параметрах.
Равновесие S ⇄ Р описывается константой равновесия Кeq (или просто К; с. 90). В стандартных условиях, в которых принято сравнивать биологические процессы, константа равновесия определяется как К'eq (или К'):
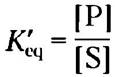 (6-2)
(6-2)
На основании законов термодинамики К'eq, и ∆G'° связаны следующим соотношением:
∆G'° = -RTInK' (6-3)
где R — универсальная газовая постоянная (8,315 ДжДмоль • К)), Т — абсолютная температура (298 К, что соответствует 25 °С). Уравнение 6-3 выводится и подробно обсуждается в гл. 13. Сейчас важно понять, что константа равновесия напрямую связана со стандартной свободной энергией реакции (табл. 6-4). Большое по абсолютной величине отрицательное значение ∆G'° означает, что равновесие реакции сдвинуто в сторону образования продуктов реакции, но, как уже говорилось выше, это не означает, что реакция может протекать с высокой скоростью.
Таблица 6-4. Соотношение между К' и ∆G'°
K'eq |
∆G'° (кДж/моль) |
10-6 |
34,2 |
10-5 |
28,5 |
10-4 |
22,8 |
10-3 |
17,1 |
10-2 |
11,4 |
10-1 |
5,7 |
1 |
0,0 |
101 |
-5,7 |
102 |
-11,4 |
103 |
-17,1 |
Примечание: соотношение рассчитано по уравнению ∆G'° = -RTInK'eq (6-3).
Скорость любой реакции определяется концентрациями реагирующих веществ и константой скорости (r). Скорость (v) мономолекулярной реакции S —> Р представляет собой количество вещества S, реагирующего в единицу времени. Скорость реакции определяется уравнением скорости:
V = r[S] (6-4)
Скорость этой реакции зависит только от концентрации S. Это так называемая реакция первого порядка. Множитель r — константа, отражающая вероятность протекания реакции при данных условиях (pH, температура и т. д.). В данном случае к является константой скорости реакции первого порядка и измеряется в единицах, обратных времени, например, с-1. Если реакция первого порядка имеет константу скорости r, скажем, 0,03 с-1, это значит, что за 1 с 3% доступного вещества S превращается в вещество Р. Реакция с константой скорости 2000 с-1 завершится за какую-то долю секунды. Если скорость реакции определяется концентрациями двух различных веществ или если реакция протекает между двумя молекулами одного и того же вещества, то такая реакция имеет второй порядок. В этом случае r является константой скорости реакции второго порядка и измеряется в л • моль-1 • с-1. Уравнение для скорости такой реакции имеет следующий вид:
V = r[S1] [S2] (6-5)
Из теории переходного состояния следует, что значение константы скорости связано с энергией активации следующим уравнением:
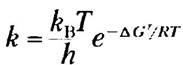 (6-6)
(6-6)
где rH — постоянная Больцмана, h — постоянная Планка. Здесь важно обратить внимание на то, что константа скорости реакции r связана с энергией активации (∆G*). В упрощенном виде это уравнение означает, что снижение энергии активации приводит к росту скорости реакции.
Теперь перейдем от вопроса, что делают ферменты, к вопросу, как они это делают.
Некоторые принципы, объясняющие высокую каталитическую активность и специфичность ферментов
Ферменты являются чрезвычайно мощными катализаторами. Они повышают скорости процессов на 5-17 порядков (табл. 6-5). Кроме того, ферменты действуют очень специфично, легко отличая свой субстрат от довольно близких по структуре соединений. Как объяснить такие большие скорости процессов при такой высокой селективности? Какой источник энергии позволяет так сильно снизить энергию активации гой или иной реакции?
Таблица 6-5. Ускорение химических реакций в присутствии ферментов
Циклофилин |
~ 105 |
Карбоангидраза |
~ 107 |
Триозофосфатизомераза |
~ 105 |
Карбоксипептидаза А |
~ 1011 |
Фосфоглюкомутаза |
~ 1012 |
Сукцинил-СоА-трансфераза |
~ 1013 |
Уреаза |
~ 1014 |
Оротидилатдекарбоксилаза |
~ 1017 |
Ответ на эти вопросы следующий. Во- первых, ферментативная реакция сопряжена с перестройками ковалентных связей. Между субстратом и функциональными группами фермента (боковыми цепями аминокислотных остатков, ионами металла, коферментами) происходят химические реакции самого разного типа. Функциональные группы фермента могут образовывать кратковременные ковалентные связи с субстратом и активировать его для химического превращения; определенная группа может переноситься с субстрата на фермент. Во многих случаях эти превращения происходят исключительно в активном центре фермента. Ковалентные взаимодействия между ферментом и субстратом ускоряют реакцию, поскольку проводят ее по другому пути, характеризующемуся более низкой энергией активации. Специфические типы изменений, происходящих в реагирующих молекулах, описаны в разд. 6.4.
Во-вторых, между ферментом и субстратом существуют и нековалентные взаимодействия. Эти слабые взаимодействия в значительной степени являются источником энергии, которая требуется для снижения энергии активации. Именно образование специфического комплекса ЕS отличает ферменты от всех прочих катализаторов. Связь фермента с субстратом в этом комплексе осуществляется при помощи тех же сил, которые стабилизируют структуру белка, а именно водородных связей, гидрофобных и ионных взаимодействий (гл. 4). Каждое слабое взаимодействие в комплексе ЕS сопровождается высвобождением небольшого количества энергии, обеспечивающей устойчивость этого взаимодействия. Энергия взаимодействия фермента с субстратом носит название энергии связывания (∆GВ). Значение этой энергии выходит далеко за пределы стабилизации фермент-субстрагного комплекса. Энергия связывания является основным источником свободной энергии, позволяющей снизить энергию активации ферментативной реакции.
Каким образом ферменты используют энергию нековалентного взаимодействия, объясняют два основополагающих и взаимосвязанных принципа.
1. В основе каталитической активности ферментов лежит свободная энергия, выделяющаяся при образовании множества слабых связей между ферментом и его субстратом. Эта энергия связывания определяет, как специфичность, так и скорость процесса.
2. Слабые взаимодействия наиболее благоприятны при прохождении реакции через переходное состояние; активный центр фермента комплементарен не самому субстрату, а тем переходным состояниям, через которые проходит субстрат в процессе своего превращения в продукт реакции.
Эти принципы очень важны для понимания сути ферментативного катализа, поэтому мы остановимся на них подробнее.
Слабые взаимодействия между ферментом и субстратом наиболее благоприятны в переходном состоянии
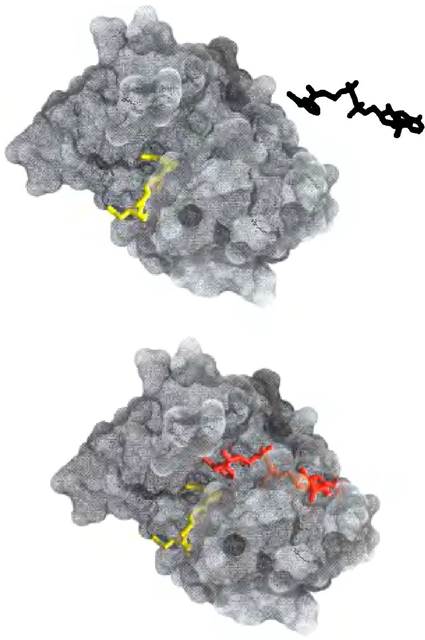
Как удается ферменту использовать энергию связывания для снижения энергии активации? Само по себе образование фермент-субстратного комплекса не дает ответа на этот вопрос, хотя ранние теории о механизмах действия ферментов строились именно на существовании этого комплекса. Изучение специфичности ферментов позволило Эмилю Фишеру в 1894 г. выдвинуть гипотезу о структурной комплементарности ферментов и их субстратов, благодаря которой они подходят друг к другу, как ключ и замок (рис. 6-4). Эта элегантная идея, утверждающая, что специфическое взаимодействие между двумя биологическими молекулами связано с комплементарностью форм их поверхностей, значительно повлияла на развитие биохимии. Выяснилось, что подобные взаимодействия лежат в основе многих биологических процессов. Однако в приложении к ферментативному катализу модель «ключа и замка» может ввести в заблуждение. Мы сейчас продемонстрируем, что фермент, который абсолютно комплементарен своему субстрату, является очень плохим катализатором.
Рис. 6-4. Комплементарность субстрата и его центра связывания на молекуле фермента. Фермент дигидрофолатредуктаза и ее субстрат NADР+ (выделен красным цветом) в свободной (наверху) и связанной формах (внизу). Еще один связанный субстрат тетрагидрофолат изображен желтым цветом (РDВ ID 1RA2). NADP+ связывается в кармане фермента, который комплементарен ему по форме и ионным свойствам. В действительности, как было показано в гл. 5, комплементарность белка и лиганда (в данном случае субстрата) редко бывает идеальной. Взаимодействие белка с лигандом часто сопровождается конформационными изменениями одной или обеих молекул (так называемое индуцированное соответствие). Отсутствие полной комплементарности фермента и субстрата играет важную роль в ферментативном катализе (на данном рисунке этого не видно).
Рассмотрим гипотетическую реакцию — разрушение намагниченного металлического стержня. На рис. 6-5, а изображена неферментативная реакция. Давайте представим себе два фермента, которые способны катализировать реакцию расщепления стержня, причем каждый из них использует магнитные силы в качестве аналога энергии связывания, используемой реальными ферментами. Сначала представим себе фермент, абсолютно комплементарный субстрату (рис. 6-5, б). Активный центр этого фермента — некий карман, выстланный магнитными частицами. Для осуществления реакции (расщепления стержня) стержень должен достичь переходного состояния, но он настолько плотно прилегает к активному центру, что не может изогнуться, поскольку изгиб приведет к исчезновению некоторых магнитных взаимодействий между стержнем и ферментом. Такой фермент мешает протеканию реакции, так как стабилизирует субстрат. Энергетическая диаграмма этой реакции (рис. 6-5, б) характеризуется наличием глубокого минимума энергии при образовании комплекса ЕS, из которого субстрат вряд ли сможет выйти. Подобный фермент абсолютно бесполезен.
Рис. 6-5. Гипотетический фермент, катализирующий разлом металлического стержня. а) Прежде чем стержень сломается, он должен изогнуться (достичь переходного состояния). В случае двух гипотетических ферментов роль слабых взаимодействий между ферментом и субстратом играют магнитные взаимодействия. б) Фермент с выстланным магнитными частицами карманом, комплементарным по форме железному стержню (субстрату), стабилизирует исходное состояние субстрата. Изгибу стержня мешают многочисленные магнитные взаимодействия с ферментом, в) Фермент, центр связывания которого комплементарен переходному состоянию, помогает дестабилизировать стержень и тем самым катализирует реакцию. Энергия связывания (магнитные взаимодействия) компенсирует рост свободной энергии, необходимый для сгибания стержня. На энергетических диаграммах (справа) видна разница в изменении энергии при комплементарности фермента исходному субстрату и его переходному состоянию (комплекс ЕР не рассматривается). Разница между энергиями переходного состояния катализируемой и некатализируемой реакций (∆GM) объясняется магнитными взаимодействиями между стержнем и ферментом. В том случае, когда фермент комплементарен субстрату (б), комплекс ЕS является более устойчивым и характеризуется более низкой свободной энергией в основном состоянии, чем сам субстрат. Это приводит к увеличению энергии активации.
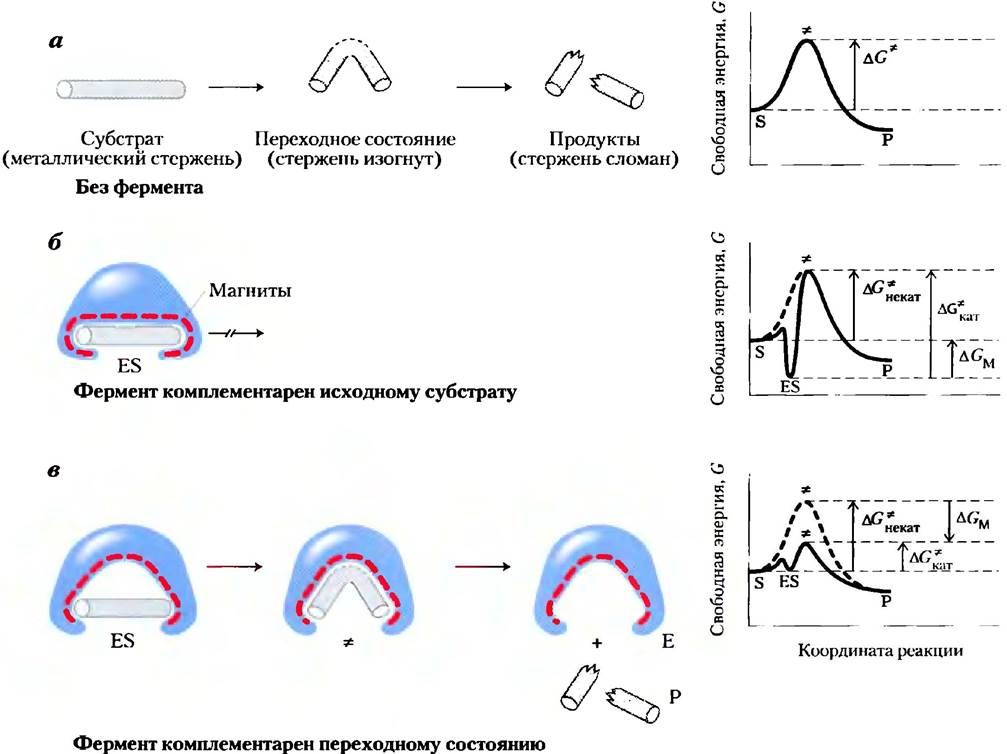
Современные представления о ферментативном катализе впервые были сформулированы Майклом Полани (1921 г.) и Джоном Холдейном (1930 г.), а затем развиты в работе Лайнуса Полинга (1946 г.). В соответствии с этой теорией, для эффективного катализа фермент должен быть комплементарен переходному состоянию реакции. Это означает, что оптимальное взаимодействие между ферментом и субстратом возможно только в переходном состоянии. На рис. 6-5, в изображен гипотетический фермент, действующий по такому принципу. Металлический стержень связывается с ферментом, но при этом реализуется только часть возможных взаимодействий. Связанный субстрат должен претерпевать дальнейшие изменения, сопровождающиеся повышением свободной энергии. Но в данном случае повышение свободной энергии, необходимое для изгиба стержня и принятия им почти изломанной конформации, возмещается за счет магнитных взаимодействий (энергии связывания) между ферментом и субстратом в переходном состоянии. Многие из этих взаимодействий происходят на участках, находящихся на значительном расстоянии от места изгиба. Эти взаимодействия между ферментом и не участвующими в реакции частями стержня обеспечивают часть энергии, необходимой для его расщепления. Такое «возмещение» энергии соответствует переходу к более низкой энергии активации и ускорению реакции.
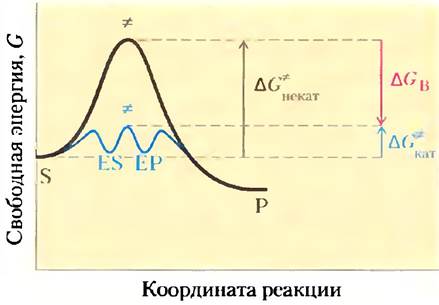
Реальные ферменты действуют по аналогичному принципу. Некоторые слабые взаимодействия существуют в фермент-субстратном комплексе, а наиболее полно эти взаимодействия реализуются, только когда субстрат достигает переходного состояния. Свободная энергия (энергия связывания), высвобождаемая при образовании этих слабых связей, частично возмещает энергию, необходимую для достижения вершины энергетического пика. Если суммировать неблагоприятный (положительный) вклад энергии активации (∆G*) и благоприятный (отрицательный) вклад энергии связывания (∆GВ), то в результате мы получим общее снижете энергии активации (рис. 6-6). Даже в случае ферментативной реакции в переходном состоянии не образуется устойчивое химическое соединение; это лишь короткий промежуток времени, когда субстрат находится в точке, соответствующей энергетическому максимуму. Следовательно, ферментативная реакция протекает гораздо быстрее неферментативной, поскольку ее энергетический барьер гораздо ниже. Именно слабое взаимодействие между ферментом и субстратом является движущей силой ферментативного катализа. Группы субстрата, участвующие в этих слабых взаимодействиях, могут находиться на отдаленных расстояниях от связей, которые разрываются или как-либо изменяются. Основной вклад в катализ вносят те слабые взаимодействия, которые возникают только в переходном состоянии.
Рис. 6-6. Роль энергии связывания в катализе. Чтобы снизить энергию активации определенной реакции, система должна получить энергию, эквивалентную снижению ∆G*. Большая часть этой энергии поступает от энергии связывания (∆G'B), появляющейся в результате образования слабых нековалентных связей между ферментом и субстратом в переходном состоянии. Роль ∆G*B в данном случае аналогична роли ∆GM на рис. 6-5.
Необходимость множества слабых взаимодействий является одной из причин, почему ферменты (и некоторые коферменты) имеют такие большие размеры. Фермент должен иметь функциональные группы для ионных, водородных связей и т. д., причем эти группы должны быть определенным образом расположены, чтобы энергия связывания была оптимальной именно в переходном состоянии. Правильное связывание обычно достигается, если субстрат попадает в полость в молекуле фермента (активный центр), где он удален от контакта с водой. Размеры белков позволяют сохранять такую структуру, в которой действующие группы расположены оптимальным образом, а активный центр поддерживается в определенном состоянии.
Энергия связывания определяет специфичность и скорость катализа
Можно ли количественно показать, что энергия связывания приводит к столь большому ускорению реакции при участии фермента? Да, можно. В качестве исходной точки используя уравнение 6-6, можно подсчитать, что в нормальных для клетки условиях для ускорения реакции первого порядка в 10 раз значение ∆G* должно уменьшиться примерно на 5,7 кДж/моль. Выигрыш в энергии при образовании одной слабой связи обычно находится в пределах от 4 до 30 кДж/моль. Таким образом, суммарной энергии, накапливающейся в результате нескольких подобных взаимодействий, вполне достаточно для снижения энергии активации на 60-100 кДж/моль, что и объясняет чрезвычайно эффективное ускорение реакций иод действием ферментов.
Та же энергия связывания, что обеспечивает энергию для катализа, определяет и специфичность фермента, т. е. его способность отличать субстрат от других молекул. Интуитивно специфичность легко отличить от катализа (ускорения реакции), гораздо труднее различить их в эксперименте, поскольку катализ и специфичность являются двумя сторонами одного и того же явления. Если активный центр фермента имеет функциональные группы, расположенные таким образом, чтобы оптимально обеспечивать слабые взаимодействия с молекулой конкретного субстрата в переходном состоянии, то этот фермент не сможет с такой же эффективностью реагировать с какой-либо другой молекулой. Например, если в молекуле субстрата есть гидроксильная группа, способная образовывать водородную связь с конкретным остатком глутаминовой кислоты (Glu) в молекуле фермента, то любая молекула, не имеющая гидроксильной группы в этом положении, будет плохим субстратом для данного фермента. Кроме того, любая молекула с функциональной группой, для которой у фермента нет места в центре связывания, по всей вероятности, не будет образовывать необходимого комплекса с ферментом. В общем, специфичность является результатом образования множества слабых связей между ферментом и его субстратом.
Значение энергии связывания для катализа легко продемонстрировать. Например, гликолитический фермент триозофосфатизомераза катализирует взаимные превращения глицеральдегид-3-фосфата и дигидроксиацетонфосфата:
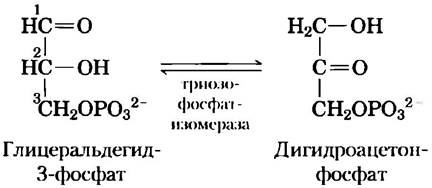
В данной реакции меняются местами карбонильная и гидроксильная группы у атомов углерода 1 и 2. Однако выяснилось, что ускорение этой ферментативной реакции более чем на 80% связано с фермент-субстратными взаимодействиями, затрагивающими фосфатную группу у атома углерода 3 в молекуле субстрата. Это удалось обнаружить при тщательном сравнении ферментативных реакций, в которых в качестве субстрата использовали глицеральдегид-3-фосфат и глицеральдегид, не имеющий фосфатной группы в положении 3.
Описанные выше принципы могут быть проиллюстрированы примерами множества известных каталитических механизмов. Эти механизмы не являются взаимоисключающими, и в действии конкретного фермента могут соединяться различные типы катализа.
Определим, что необходимо для протекания реакции. Заметными физическими и термодинамическими факторами, определяющими энергетический барьер реакции (∆С*), могут быть: 1) энтропия (мера беспорядка) молекул в растворе, которая уменьшает возможность их взаимодействия; 2) сольватная оболочка из связанных водородными связями молекул воды, окружающая и стабилизирующая большинство макромолекул в водном растворе; 3) искажение молекулы субстрата, происходящее во многих реакциях; 4) необходимость определенной организации функциональных групп фермента. Энергия связывания расходуется на преодоление всех этих барьеров.
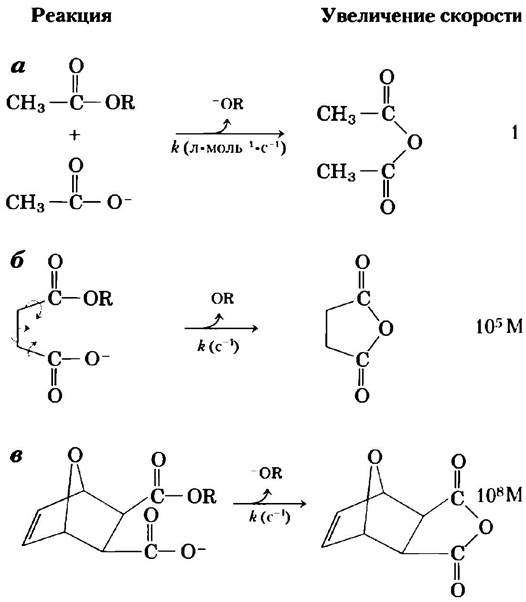
Во-первых, очевидным положительным результатом связывания двух реагирующих субстратов с ферментом является значительное ограничение их подвижности, выражающееся в снижении энтропии. Энергия связывания удерживает субстраты в ориентации, необходимой для протекания реакции, что вносит ощутимый вклад в катализ, поскольку продуктивные столкновения молекул в растворе чрезвычайно редки. Субстраты таким образом размещаются на молекуле фермента, чтобы множество слабых взаимодействий между ними и стратегическими группами фермента удерживало молекулы субстратов в необходимом положении. Исследования показали, что ограничение подвижности двух реагирующих веществ на несколько порядков увеличивает скорость реакции (рис. 6-7).
Рис. 6-7. Увеличение скорости реакции в результате снижения энтропии. Здесь показана реакция между сложным эфиром и карбоксильной группой, приводящая к образованию ангидрида. В каждом случае R-группа одна и та же. а) Данная бимолекулярная реакция описывается константой скорости реакции второго порядка, измеряемой в л • моль-1 • с-1, б) Если обе реагирующие группы находятся на одной молекуле, реакция протекает гораздо быстрее. Константа к этой мономолекулярной реакции измеряется в с-1. Разделив значение константы скорости реакции (б) на константу скорости реакции (а), получим увеличение скорости около 105 М (увеличение скорости имеет размерность, поскольку мы сравниваем мономолекулярную и бимолекулярную реакции). Иными словами, если реагирующее вещество в реакции (б) находится в концентрации 1 М, то реакция проходит так, как будто концентрация реагирующих групп составляет 105 М. Заметьте, что реагирующая молекула в реакции (б) имеет свободу вращения вокруг трех связей (показаны стрелками); тем не менее в этом случае наблюдается значительное снижение энтропии по сравнению с реакцией (а). В случае (в) все те связи, что допускали вращение в случае (б), жестко фиксированы. В результате энтропия еще больше снижается, а реакция по сравнению со случаем (а) ускоряется в 108 раз.
Во-вторых, образование слабых связей между субстратом и ферментом приводит к разрушению сольватной оболочки вокруг субстрата. Связи фермента с субстратом замещают большинство водородных связей между субстратом и водой.
В-третьих, энергия связывания, являющаяся результатом слабых взаимодействий, возникающих исключительно в переходном состоянии, помогает компенсировать потери от любого искажения молекулы, в первую очередь от перераспределения электронов, которому субстрат должен подвергнуться в реакции.
И наконец, в-четвертых, при связывании субстрата сам фермент обычно претерпевает конформационные изменения, что связано с образованием множества слабых связей. Данный процесс называют индуцированным соответствием, механизм которого был предложен Д. Кошландом в 1958 г. Эти сдвиги могут происходить в небольшой части молекулы фермента около активного центра, но могут также затрагивать целые домены. Обычно в молекуле фермента происходит целая серия связанных между собой перемещений, которые в конечном итоге приводят к необходимым изменениям состояния активного центра. Индуцированное соответствие приводит к той ориентации функциональных групп фермента, которая необходима для осуществления катализа. Конформационные изменения также способствуют образованию в переходном состоянии дополнительных слабых связей. В любом случае новая конформация фермента имеет улучшенные каталитические свойства. Раньше мы уже показали, что индуцированное соответствие является общим принципом обратимого связывания белков с лигандами (гл. 5); оно также играет важную роль во взаимодействии практически любого фермента с его субстратом.
Роль специфических каталитических групп в катализе
Для большинства ферментов энергия связывания, используемая для образования комплекса ЕS, является лишь частью той движущей силы, которая способствует катализу. Как только субстрат связывается с ферментом, определенным образом расположенные функциональные группы помогают расщеплять и образовывать связи, действуя в соответствии с различными механизмами, в том числе общего кислотно-основного катализа, ковалентного катализа и катализа ионами металлов. Эти механизмы отличны от тех, что используют энергию связывания, поскольку в данном случае происходит кратковременное образование ковалентных связей с субстратом или перенос группы от субстрата или на него.
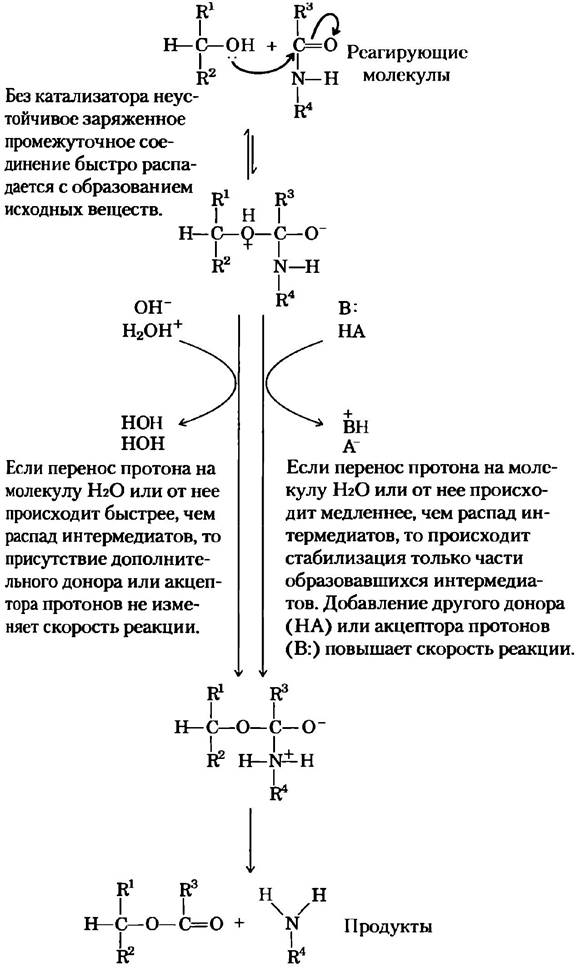
Общий кислотно-основной катализ. Многие биохимические реакции проходят через стадию образования неустойчивых заряженных интермедиатов, которые быстро распадаются на исходные соединения и тем самым препятствуют протеканию реакции (рис. 6-8). Заряженные интермедиаты часто можно стабилизировать путем переноса протонов от субстрата (интермедиата) или на него, в результате чего образуется соединение, которое легче распадается с образованием продуктов реакции. В неферментативных реакциях в переносе протона участвуют либо только компоненты молекулы воды, либо другие слабые доноры и акцепторы протонов. Катализ такого типа, который использует исключительно Н+ (Н3O+) или ОН-, присутствующие в воде, называется специфическим кислотно-основным катализом. Если перенос протонов между интермедиатом и водой происходит быстрее, чем интермедиат распадается на исходные вещества, то он эффективно стабилизируется сразу после образования. Никакого дополнительного катализа посредством других доноров или акцепторов протонов не происходит. Однако во многих случаях участия воды недостаточно. Общим кислотно-основным катализом называют перенос протонов, осуществляемый другими классами молекул. В неферментативных реакциях в водных растворах этот вариант катализа наблюдается в том случае, если неустойчивый интермедиат распадается на исходные вещества быстрее, чем происходит перенос протона на молекулу воды или от нее. В данной ситуации многие слабые органические кислоты могут служить дополнительными донорами протонов, а слабые органические основания акцепторами протонов.
Рис. 6-8. Преодоление неблагоприятного образования заряда в процессе расщепления амидной связи. Реакция, аналогичная изображенному здесь гидролизу амидной связи, происходит при катализе химотрипсином и другими протеазами. Образование заряда является неблагоприятным фактором, и он должен быть скомпенсирован путем присоединения протона от Н3O+ (специфический кислотный катализ) или любой кислоты НА (общий кислотный катализ). Кроме того, заряд может быть нейтрализован переносом протона на ОН- (специфический основной катализ) или основание В (общий основной катализ).
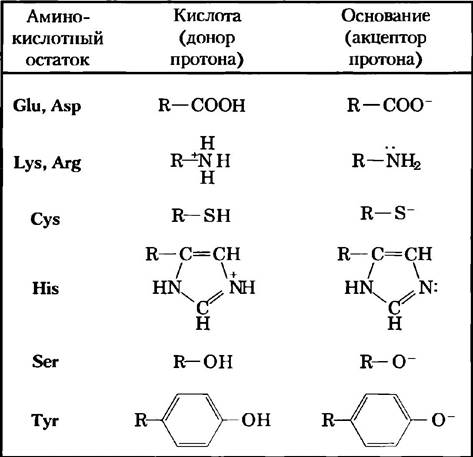
Аналогичным образом, в активном центре фермента боковые цепи ряда аминокислотных остатков могут играть роль доноров и акцепторов протонов (рис. 6-9). В активном центре фермента эти группы располагаются определенным образом, чтобы осуществлять перенос протона, и обеспечивают увеличение скорости реакции до 102 — 105 раз. В соответствии с этим механизмом катализа действует большинство ферментов. Перенос протона относится к разряду наиболее распространенных биохимических реакций.
Рис. 6-9. Аминокислоты, участвующие в общем кислотно-основном катализе. Многие органические реакции протекают при участии доноров протонов (кислот) или акцепторов протонов (оснований). Активные центры некоторых ферментов содержат функциональные группы аминокислот, в частности перечисленные здесь, которые могут принимать участие в процессе катализа в качестве доноров или акцепторов протонов.
Ковалентный катализ. При ковалентном катализе между ферментом и субстратом временно образуется ковалентная связь. Рассмотрим реакцию гидролиза связи между группами А и В:
![]()
В присутствии ковалентного катализатора (фермента с нуклеофильной группой X:) реакция приобретает следующий вид:
![]()
Таким образом, путь реакции меняется, и катализ осуществляется, только если энергия активации нового пути ниже, чем энергия активации некаталитической реакции. Обе стадии нового процесса должны протекать быстрее, чем реакция без катализатора. В качестве нуклеофильных групп, участвующих в образовании ковалентных связей с субстратом, могут выступать боковые цепи некоторых аминокислотных остатков, как те, что показаны на рис. 6-9, а также некоторые кофакторы. После образования ковалентного комплекса обязательно происходит реакция, в результате которой высвобождается свободный фермент. Ковалентные связи между ферментом и субстратом могут специфическим образом активировать субстрат для последующих реакций.
Катализ ионами металлов. Металлы, как прочно связанные с ферментом, так и захватываемые из раствора вместе с субстратом, могут участвовать в катализе несколькими способами. Ионные взаимодействия между субстратом и связанным с ферментом металлом помогают ориентировать субстрат определенным образом или стабилизировать заряженную молекулу субстрата в переходном состоянии. Такое использование слабых взаимодействий между металлом и субстратом напоминает некоторые типы использования энергии связывания фермента с субстратом, описанные ранее. Кроме того, металлы могут способствовать протеканию окислительно-восстановительных реакций, обратимо изменяя свою степень окисления. Около трети известных на сегодняшний день ферментов для проявления каталитической активности нуждаются в присутствии одного или нескольких ионов металлов.
Большинство ферментов совмещают несколько стратегий катализа. Хорошим примером одновременного использования ковалентного, общего кислотно-основного катализа и стабилизации в переходном состоянии может служить реакция, катализируемая химотрипсином, которая более подробно рассматривается в разд. 6.4.
Краткое содержание раздела 6.2 Как работают ферменты
■ Ферменты — это исключительно активные катализаторы, ускоряющие реакции, как правило, в 105 — 1017 раз.
■ Ферментативные реакции характеризуются образованием фермент-субстратного комплекса (ЕS). Связывание субстрата происходит в полости молекулы фермента, называемой активным центром.
■ Суть действия ферментов и других катализаторов заключается в снижении энергии активации (∆С*), в результате чего увеличивается скорость реакции. Катализатор не смещает равновесия реакции.
■ Значительная доля энергии, идущей на повышение скорости реакции, возникает за счет слабых взаимодействий (водородных, гидрофобных, ионных) между ферментом и субстратом. Активный центр фермента устроен таким образом, что некоторые из этих слабых взаимодействий реализуются только в переходном состоянии и стабилизируют его. Необходимость множества слабых связей является одной из причин, объясняющих большие размеры молекул ферментов. Энергия связывания фермента с субстратом (∆GB) используется для снижения энтропии субстрата или осуществления конформационных изменений в молекуле фермента (индуцированное соответствие). Энергия связывания, кроме того, вносит вклад в специфичность ферментов к их субстратам.
■ К дополнительным каталитическим механизмам с участием ферментов относятся общий кислотно-основной катализ, ковалентный катализ и катализ ионами металлов. Ферменты часто образуют временные ковалентные связи с субстратами или осуществляют перенос определенных групп, в результате чего возникает новый путь реакции с более низкой энергией активации.