Биохимия человека Том 1 - Марри Р. 1993
Биоэнергетика и метаболизм углеводов и липидов
Метаболизм наиболее важных гексоз
Метаболизм галактозы
Галактоза образуется при гидролизе в кишечнике дисахарида лактозы (молочного сахара). В печени она легко превращается в глюкозу. Способность печени осуществлять это превращение может быть использована в качестве функциональной пробы — теста на толерантность к галактозе. Путь превращения галактозы в глюкозу показан на рис. 21.3.
Галактоза фосфорилируется в результате реакции 1, катализируемой галактокиназой (донором фосфата служит АТР). Продукт реакции — галактозо-1-фосфат реагирует с уридиндифосфат-глюкозой (UDP-глюкозой) с образованием уридин-дифосфатгалактозы (UDP-галактозы) и глюкозо-1-фосфата. На этой стадии (реакция 2), катализируемой ферментом галактозо-1-фосфат-уридил-трансферазой, галактоза занимает место глюкозы в UDP-глюкозе с образованием UDP-галактозы. Превращение галактозы в глюкозу (реакция 3) происходит в составе галактозосодержащего нуклеотида. Эта реакция, продуктом которой является UDP-глюкоза, катализируется эпимеразой. Реакция эпимеризации, вероятно, включает стадии окисления и восстановления по С-4 с участием NAD в качестве кофермента. Наконец, глюкоза высвобождается из UDP-глюкозы в виде глюкозо-1-фосфата (реакция 4), возможно, после включения в гликоген и последующего его фосфоролиза.
Реакция 3 легко обратима, таким путем глюкоза может превращаться в галактозу, и последняя, следовательно, не является незаменимым компонентом пищи. Галактоза необходима для образования не только лактозы, но и гликолипидов (цереброзидов), протеогликанов и гликопротеинов.
При синтезе лактозы в молочной железе сначала из глюкозы и нуклеотида при участии вышеперечисленных ферментов образуется UDP-галактоза. Затем она вступает в катализируемую лактозосинтазой реакцию с глюкозой, в результате которой образуется лактоза.
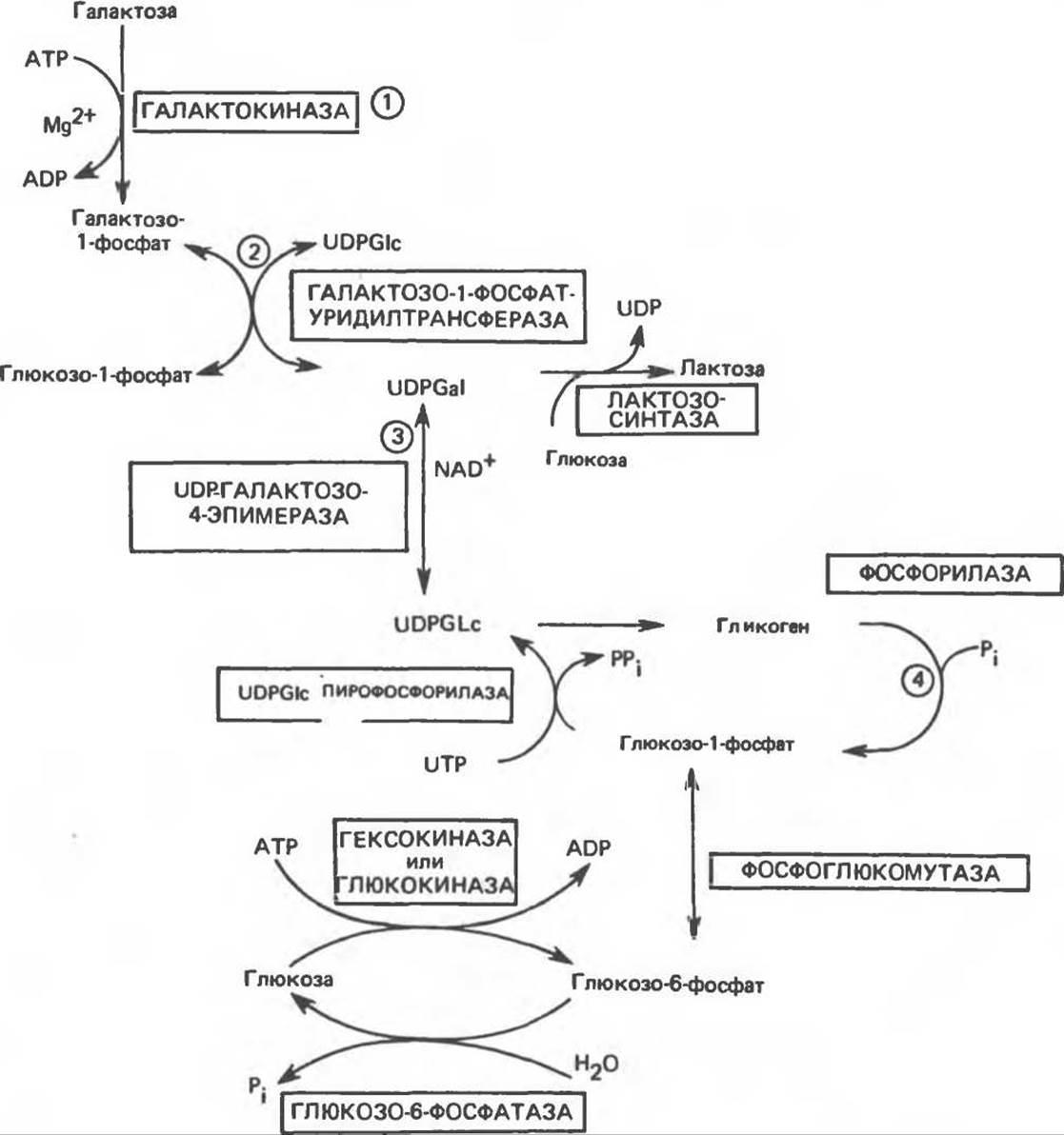
Рис. 21.3. Путь превращения галактозы в глюкозу и путь синтеза лактозы.
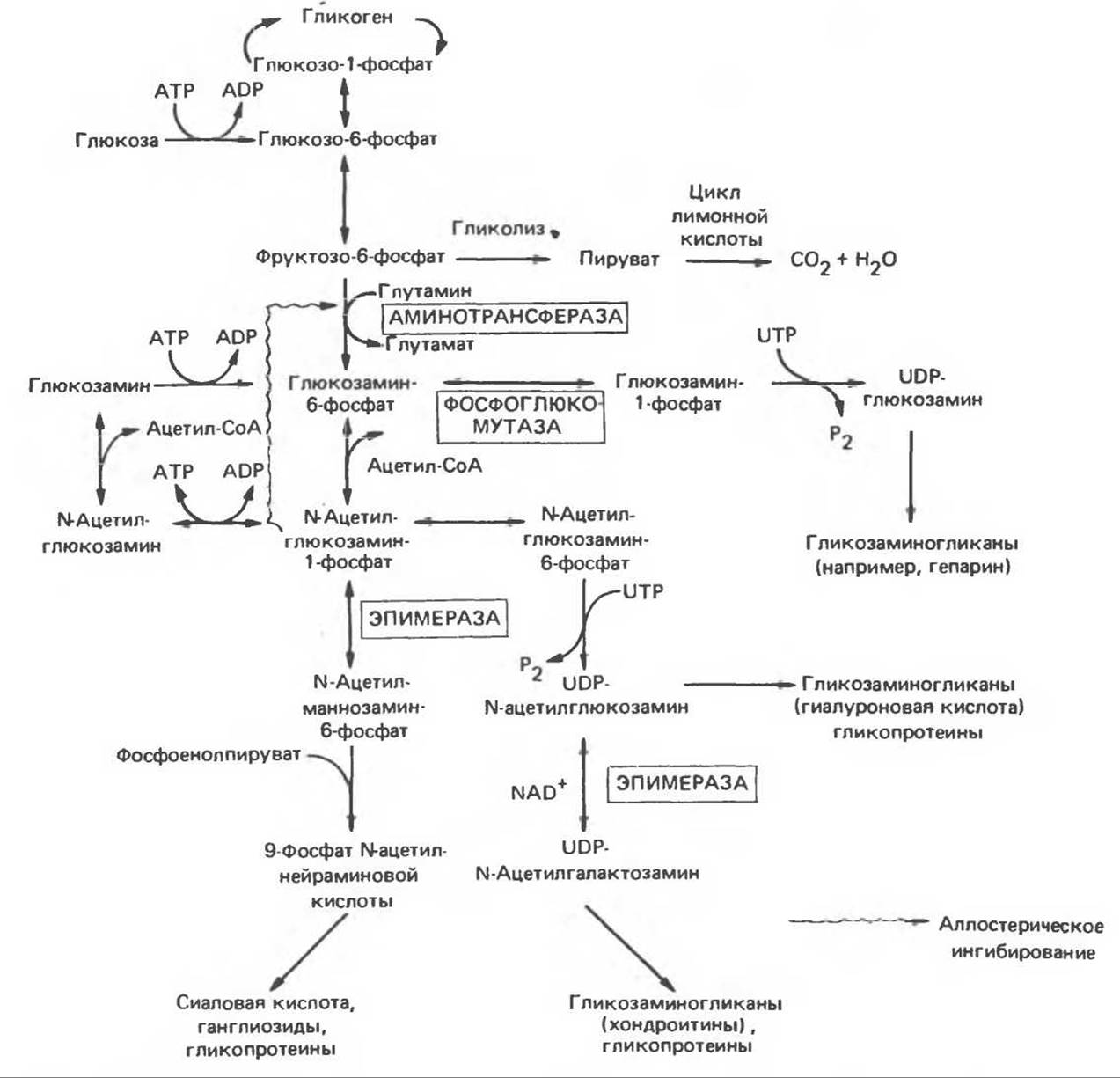
Рис. 21.4. Схема взаимосвязи метаболизма аминосахаров. UDP-глюкозамин — аналог UDP-Glc. Другие пуриновые или пиримидиновые нуклеотиды могут аналогично связывать сахара или аминосахара. Примерами таких соединений могут служит TDP-глюкозамин и TDP-N-ацетилглюкозамин.
Клинические аспекты
Нарушение метаболизма галактозы наблюдается при галактоземии, которая может быть вызвана наследственными дефектами в любом из трех ферментов, обозначенных 1, 2, 3 на рис. 21.3. Наиболее хорошо известным является недостаток уридилтран-сферазы (2). При увеличении концентрации галактозы в крови повышается ее концентрация в тканях. В тканях глаза она восстанавливается альдозоредуктазой с образованием соответствующего полиола (галактитола). Накопление галактитола способствует развитию катаракты. Весьма тяжелые последствия наблюдаются при дефиците уридилтрансферазы: в печени происходит накопление галактозо-1-фосфата, при этом соответственно снижается концентрация неорганического фосфата. В результате возникает нарушение функции печени, а затем расстройство психики.
Если при наследственном дефиците галактозо-1-фосфат-уридилтрансферазы (реакция 2), приводящем к нарушению метаболизма галактозы в печени и красных кровяных тельцах, эпимераза (реакция 3) присутствует в достаточном количестве, то у больных может происходить образование UDP-галактозы из глюкозы. Это объясняет, почему дети с таким заболеванием могут нормально расти и развиваться при назначении диеты, из которой исключена галактоза (такая диета назначается для предотвращения тяжелых форм заболевания). Описано несколько различных генетических дефектов, которые вызывают не полный, а частичный дефицит трансферазы. Поскольку обычно этот фермент присутствует в организме в избытке, снижение его активности до 50% (и даже ниже) может не сопровождаться клиническими проявлениями заболевания; последние наблюдаются у гомозиготных индивидуумов. В тех случаях, когда имеется дефицит эпимеразы в эритроцитах, при наличии данного фермента в печени и других органах симптомы заболевания не обнаруживаются.
Метаболизм аминосахаров (гексозаминов) (рис. 21.4)
Аминосахара являются важными компонентами гликопротеинов (см. гл. 54), некоторых гликосфинголипидов (например, ганглиозидов, см. гл. 15) и гликозаминогликанов (см. гл. 54). Наибольшее значение среди них имеют глюкозамин, галактозамин, маннозамин (все они — гексозамины) и С-9-соединение — сиаловая кислота. Главной сиаловой кислотой, обнаруженной в тканях человека, является N-ацетилнейраминовая кислота (NeuAc). Схема реакций взаимных превращений аминосахаров представлена на рис. 21.4; наиболее существенные моменты ее следующие: (1) главным аминосахаром является глюкозамин; он образуется из фруктозо-6-фосфата в форме глюкозамин-6-фосфата, причем донором аминогруппы является глутамин; (2) аминосахара функционируют в основном в N-ацетилированной форме, донором ацетила является ацетил-СоА; (3) N-ацетилманнозамин-6-фосфат образуется путем эпимеризации N-ацетилглюкозамин-6-фосфата; (4) NeuAc образуется в результате конденсации маннозамин-6-фосфата с фосфоенолпируватом; (5) галактозамин образуется путем эпимеризации UDP-N-ацетилглюкозамина (UDPGlcNAc) в UDP-N-ацетилгалактозамин (UDP-GalNAc); (6) аминосахара используются для биосинтеза гликопротеинов и других соединений в форме нуклеотидсахаров, основными из которых являются UDPGlcNAc, UDPGalNAc и CMPNeuAc.
Литература
Brown D.H., Brown В. I. Some inborn errors of carbohydrate metabolism. Page 391. In: MTP International Review of Science, Vol 5, Whelan W. J. (ed.), Butterworth, 1975.
Dickens F., Randei P.J., Whelan W.J. (ed.). Carbohydrate Metabolism and Its Disorders, 2 vols, Academic Press, 1968. Huijing F. Textbook errors, Galactose metabolism and galactosemia, Trends Biochem. Sсi., 1978, 3, N 129.
James H. M. et al. Models for the metabolic production of oxalate from xylitol in humans, Aust. J. Exp. Biol. Med. Sсi., 1982, 60, 117.
Kador P. F., Akagi Y., Kinoshita J. H. The effects of aldose reductase and its inhibition on sugar cataract formation, Metabolism, 1986, 35, 15.
Macdonald I., Vrana A. feds). Metabolic Effects of Dietary Carbohydrates, Karger, 1986.
Randle P. J., Steiner D.F., Whelan W.J. (eds). Carbohydrate Metabolism and Its Disorders, Vol 3, Academic Press, 1981.
Sperling O., de Vries A. (eds). Inborn Errors of Metabolism in Man, Karger, 1978.
Stanbury J. B. et al. (eds). The Metabolic Basis of Inherited Disease, 5th ed. McGraw-Hill, 1983.