Биохимия человека Том 2 - Марри Р. 1993
Частные вопросы
Плазма крови и процесс свертывания
Свертывание крови
Прекращение кровотечения после травматического повреждения кровеносных сосудов называется гемостазом. Выделяют четыре фазы гемостаза: первая фаза — сокращение поврежденного сосуда. При этом уменьшается кровоснабжение дистальной от травмы области. Вторая фаза — образование в месте повреждения рыхлой тромбоцитарной пробки или белого тромба. Имеющийся в участке повреждения коллаген служит связывающим центром для тромбоцитов; у последних в результате связывания разрушается их внутренняя структура и высвобождаются тромбоксан и ADP. Они в свою очередь индуцируют присоединение новых тромбоцитов и таким образом образуется рыхлая временная пробка. Длительность данной фазы гемостаза определяют по продолжительности кровотечения. Третья фаза — формирование красного тромба (кровяного сгустка). Четвертая фаза — частичное или полное растворение сгустка.
Различают три типа тромбов или сгустков. Белый тромб образуется из тромбоцитов и фибрина; в нем относительно мало эритроцитов. Формируется он в местах повреждения или на патологически измененной стенке сосуда в условиях высокой скорости кровотока (в артериях). Второй вид тромбов — это диссеминированные отложения фибрина в очень мелких сосудах (капиллярах).
Третий вид тромбов — красный тромб — состоит из эритроцитов и фибрина. Морфология красного тромба сходна с морфологией сгустков, образующихся в пробирке. Красные тромбы формируются in vivo в областях замедленного кровотока при отсутствии патологических изменений в стенке сосуда, в месте повреждения или на измененной стенке сосу
да вслед за инициирующей тромбоцитарной пробкой. Инициация образования сгустка в ответ на повреждение ткани осуществляется по внешнему пути свертывания. Инициация формирования красного тромба в области замедленного кровотока или на аномальной сосудистой стенке при отсутствии повреждения ткани происходит по внутреннему пути свертывания. Внешний и внутренний пути свертывания завершаются общим конечным путем. На этом этапе происходит переход протромбина в тромбин и катализируемое тромбином превращение фибриногена в фибрин тромба.
Превращение фибриногена в фибрин, катализируемое тромбином
Фибриноген1 (фактор 1, см. рис. 55.1 и табл. 55.3) — это растворимый гликопротеин плазмы, синтезируемый в печени; длина его молекулы 46 нм, мол. масса 340 000. Молекула состоит из шести полипептидных цепей (две Aa-цепи, 2Вβ-цепи и две у-цепи). Структура фибриногена — Aa2Bß2y2. Bß- и уцепи содержат сложные олигосахариды, связанные с остатками Asn. Все три гена, кодирующие Aa-, Bß- и у-цепи, сцеплены; их активность у человека координированно регулируется. Концы молекул фибриногена обладают сильным отрицательным зарядом; это обусловлено присутствием большого количества остатков аспартата и глутамата в A-области цепи Аа и В-области цепи Bß (рис. 55.6). Помимо этого В-область цепи Bß содержит необычный отрицательно заряженный остаток тирозин-О-сульфата. Отринательно заряженные концы молекул фибриногена не только способствуют растворимости последних в воде, они отталкивают концы других молекул фибриногена, что предотвращает агрегацию последних.
Таблица 55.3. Система нумерации факторов свертывания крови. Номера не отражают последовательности действия факторов
|
Фактор |
Название |
|
I |
Фибриноген |
|
II |
Протромбин |
|
IV |
Кальций |
|
V |
Лабильный фактор, проакселерин, Ас-глобулин |
|
VII |
Проконвертин, ускоритель превращения сывороточного протромбина, котромбопластин, аутопротромбин I |
|
VIII |
Антигемофильный фактор, антигемофильный глобулин |
|
IX |
Тромбопластиновый компонент плазмы (фактор Кристмаса) |
|
X |
Фактор Стюарта-Провера |
|
XI |
Предшественник тромбопластина плазмы |
|
XII |
Фактор Хагемана |
|
XIII |
Фактор Лаки-Лоранда |
1 Все факторы свертывания (за исключением фибриногена, протромбина и продуктов их активации, а также ионов Са2+) обозначены римскими цифрами (табл. 55.3).
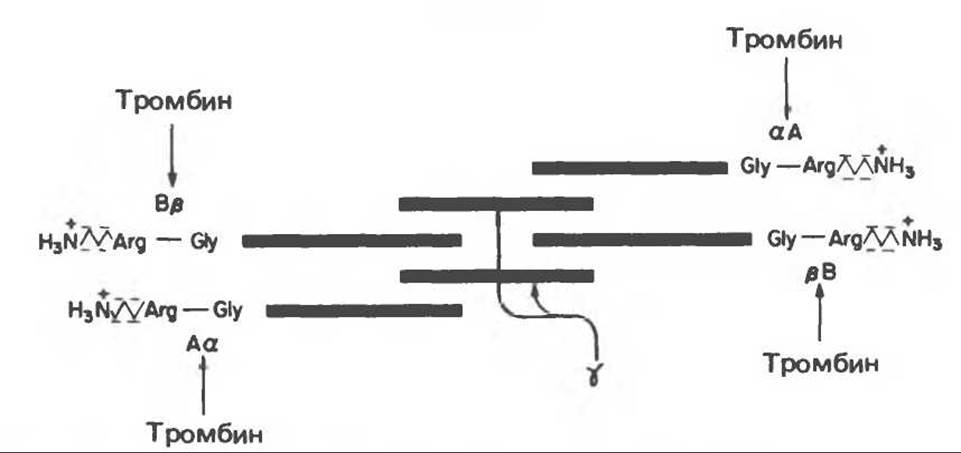
Рис. 55.6. Схематическое изображение фибриногена, его структуры (AaBßy)2, заряженных концов, сайтов расщепления тромбином (стрелки) четырех пептидных связей Arg-Gly.
Тромбин — это сериновая протеаза с мол. массой 34000, состоящая из двух полипептидных цепей. Тромбин гидролизует четыре пептидные связи Arg-Gly в фибриногене (рис. 55.6). Из этих четырех связей две соединяют области А и а, а другие две — области В и β в цепях Аа и Bß соответственно. Удаляемые из молекулы фибриногена фрагменты А и В являются отрицательно заряженными фибринопептидами, в результате образуется мономер фибрина, имеющий структуру (aßy)2. Длинные нерастворимые мономеры фибрина спонтанно ассоциируют в регулярные зигзагообразные структуры; в результате образуется нерастворимый полимерный фибриновый сгусток. Он захватывает эритроциты, тромбоциты и другие компоненты крови, в результате чего образуется красный тромб или белый тромб (тромбоцитарная пробка). На ранней стадии фибриновый сгусток представляет собой весьма рыхлое образование, удерживающееся лишь нековалентносвязанной системой нерастворимых фибриновых мономеров.
Функция тромбина помимо превращения фибриногена в фибрин заключается в переводе фактора XIII в его активную форму (ХІІІа). Фактор ХІІІа (трансглутаминаза) «сшивает» мономеры фибрина путем образования специфической изопептидной связи между у-карбоксамидной группой глутамина и Е-аминогруппой лизина (рис. 55.7). Такая стабилизация фибринового сгустка способствует его ретракции, что можно наблюдать в пробирке. Повышенная кровоточивость, наблюдаемая у пациентов с наследственной недостаточностью фактора XIII, объясняется невозможностью образования стабильного фибринового сгустка.
Известно, что внезапный тромбоз сосудов может иметь опасные и даже катастрофические последствия. Вот почему активность тромбина должна в организме тщательно контролироваться. Такой контроль осуществляется двумя механизмами. Один из них опосредован функцией антагониста тромбина — антитромбина III (см. ниже). Второй механизм состоит в том, что в организме синтезируется и циркулирует каталитически неактивный зимоген тромбина — протромбин. Протромбин, или фактор II, синтезируется в печени и содержит остатки у-карбоксиглутамата (Gla). Протромбин представляет собой одноцепочечный гликопротеин с мол. массой 72000; рис. 55.8 знакомит нас с первичной и вторичной структурой этой молекулы. N-концевая область протромбина (1 — на рисунке) содержит до 14 остатков Gla. Пунктирной линией обозначен дисульфидный мостик между областями А и В протромбина. Черным треугольником отмечена локализация каталитически активного остатка серина протеазного центра.
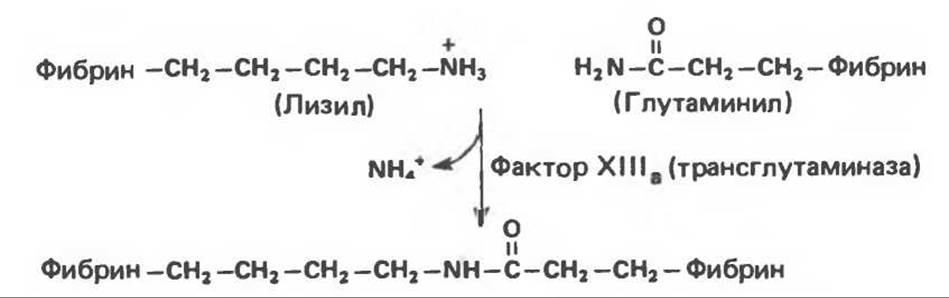
Рис. 55.7. Поперечная сшивка фибриновых молекул при действии активированного фактора XIII.
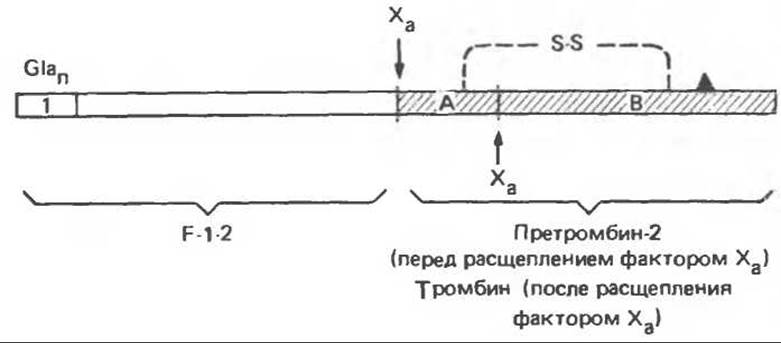
Рис. 55.8. Схематическое строение протромбина. N-конец — слева; область 1 содержит все остатки Gla Показаны сайты расщепления фактором Ха и наименования продуктов расщепления. Локализация каталитически активного остатка серина обозначена ▲. А- и В-цепи активного тромбина (заштрихованы) удерживаются вместе дисульфидным мостиком.
Активация протромбина происходит на тромбоцитах; в этом процессе участвуют анионный тромбоцитарный фосфолипид, ионы Са2+, факторы Va и Ха.
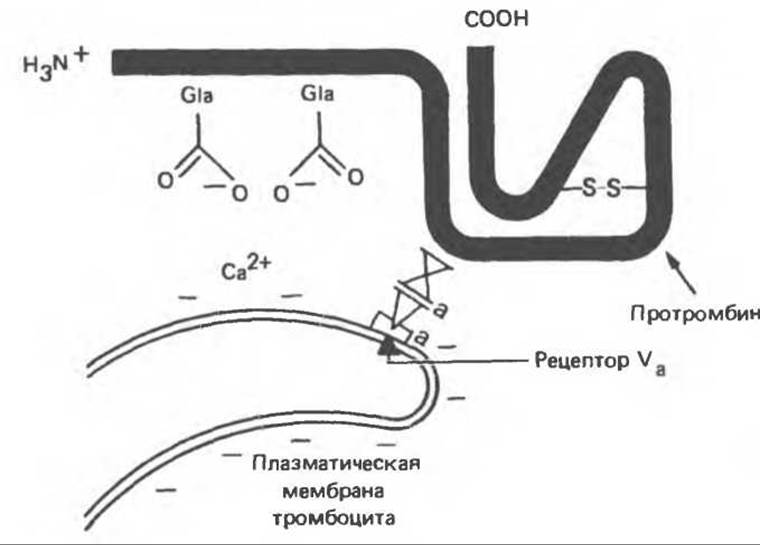
Рис. 55.9. Схема связывания факторов Va,Xa, ионов Са2+ и протромбина с плазматической мембраной тромбоцита.
Фосфолипиды, находящиеся на внутренней стороне плазматической мембраны тромбоцитов, экспонируются в результате индуцированного коллагеном разрушения и дегрануляции тромбоцитов. Эти фосфолипиды связывают ионы Са2+ и протромбин (последний, по N-концевой области, содержащей остатки Gla). Тромбоциты содержат также фактор V, который в активированной форме (Va) соединяется со специфическими рецепторами на мембране тромбоцитов (рис. 55.9). Фактор Va служит рецептором для фактора Ха, который в свою очередь связывает протромбин в области F-1∙2 (рис. 55.8). Фактор Ха также является сериновой протеазой, он расщепляет каталитически неактивную молекулу протромбина в областях, указанных на рис. 55.8. При этом высвобождается N-концевая часть протромбина. В результате расщепления тромбина фактором Ха образуются полипептиды тромбина А и В, связанные дисульфидным мостиком.
Связывание фосфолипида через ионы Са2+ с остатками Gla протромбина усиливает процесс активации последнего в 50—100 раз. Это происходит, по-видимому, вследствие создания высокой локальной концентрации протромбина и фактора Ха (рис. 55.9). Фактор Va вызывает усиление активации протромбина примерно в 350 раз также благодаря повышению локальной концентрации фактора Ха.
Фактор Va, образуемый под действием тромбина из фактора V, впоследствии тем же тромбином и инактивируется, таким путем ограничивается процесс активации протромбина в тромбин.
Протромбин может быть активирован стафилокоагулазой в результате конформационных изменений.
Активация фактора X
На этапе активации фактора X происходит соединение внешнего и внутреннего путей и образуется общий конечный путь свертывания крови (рис. 55.10). Фактор X представляет собой зимоген сериновой протеазы с мол. массой 55000 и содержит остатки Gla. Как и в протромбине, остатки Gla фактора X обеспечивают Са2+-зависимое связывание с кислыми фосфолипидами мембран тромбоцитов. Для превращения фактора X в его активную форму (X) необходимо расщепление связи аргинин-изолейцин с помощью другой сериновой протеазы. Известны две сериновые протеазы, расщепляющие связь Arg— Ilе в молекуле фактора X.
Внешний путь образования фактора Ха
Разрыв связи Arg—Іlе, а следовательно, превращение фактора X в фактор Хa. на внешнем пути осуществляют совместно фактор VIIa и тканевый фактор. Фактор VІІa функционирует только на внешнем пути, который быстро включается после повреждения ткани. Предшественник фактора VIIa — фактор VII (еще один Gla-содержащий гликопротеин) — синтезируется в печени и может активироваться тромбином или фактором Ха. Фактор VII — это зимоген, однако он обладает относительно высокой эндогенной активностью. Тканевый фактор, ускоряющий действие факторов VII или VIIa на фактор X, в большем количестве содержится в плаценте, легких и мозге.
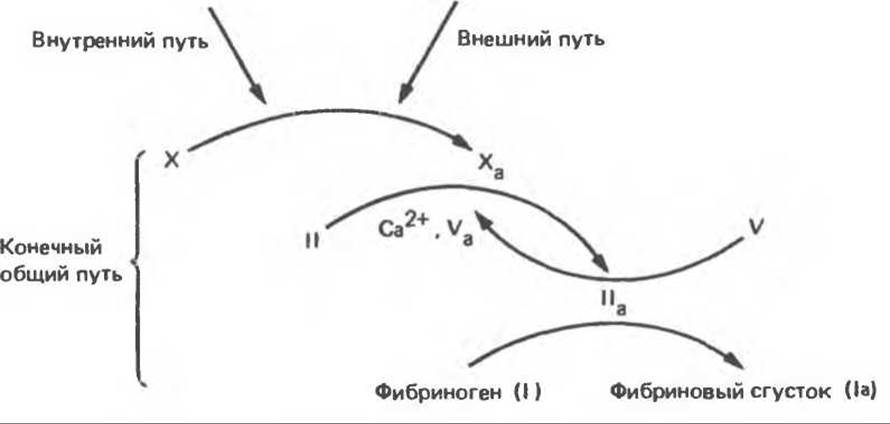
Рис. 55.10. Взаимосвязь внутреннего, внешнего и конечного общего пути в процессе свертывания крови.
В 1 мл плазмы содержится примерно 3 мг фибриногена и только 0,01 мг фактора X. Это означает, что в системе свертывания должна иметь место амплификация. И действительно, превращение фактора X в Ха — аутокаталитический процесс, который можно рассматривать как амплификацию. В рассмотренной группе реакций нелегко понять, что является первичным — «курица или яйцо»; в данном случае — фактор IIа (тромбин) или фактор Ха (рис. 55.10).
Внутренний путь образования фактора Ха
Внутренний путь образования фактора Ха начинается с взаимодействия in vivo прекалликреина, высокомолекулярного кининогена, факторов XII и XI на активирующей поверхности, вероятно на коллагене (рис. 55.11). Активирующей поверхностью внутреннего пути в опытах in vitro служит стекло и каолин. Взаимодействие фактора XII с активирующей поверхностью делает его более доступным для протеолитической атаки калликреином. В результате действия калликреина образуется фактор ХIIа, который в свою очередь индуцирует переход прекалликреина в калликреин. Таким образом, имеет место реципрокная активация. Фактор ХIIа высвобождает из высокомолекулярного кининогена брадикинин и активирует фактор XI в ХIа. Фактор ХIа в результате двух последовательных реакций активирует фактор IX (Gla-содержащий зимоген). Фактор IХа в присутствии ионов Са2+ и кислых фосфолипидов медленно активирует фактор X; активация происходит путем расщепления той же связи Arg—Ile, которую расщепляет фактор VIIa на внешнем пути. Скорость активации фактора X фактором ІХа увеличивается в 500 раз в присутствии фактора VIII (или VIIIa). Для активации фактора VIII, по-видимому, необходимо небольшое количество тромбина. Фактор VIII не является протеазой; вероятно, он служит рецептором для фактора IXа при расщеплении последним связи Arg—Ile в факторе X. Внутренний путь свертывания крови — медленный процесс, поскольку в нем участвует большое число факторов. Все вместе они образуют каскадный механизм, генерирующий фактор Ха (рис. 55.11).
В табл. 55.4 представлен целый ряд наследственных болезней человека, обусловленных недостаточностью различных компонентов системы свертывания. Наиболее часто наблюдается недостаточность фактора VIII, детерминирующая гемофилию А (соответствующий ген локализован в 10-й хромосоме человека). Эта болезнь сыграла значительную роль в истории королевских династий в Европе.
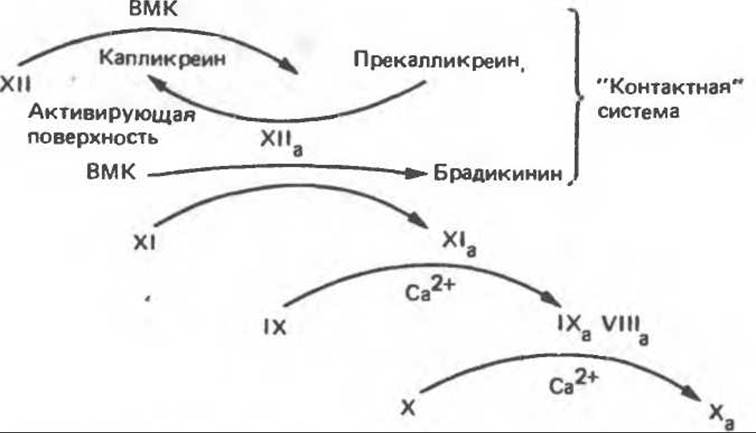
Рис. 55.11. Внутренний путь активации фактора X в Ха. ВМК — высокомолекулярный кининоген.
Таблица 55.4. Геморрагические заболевания и связанные с ними нарушения
|
Фактор |
Заболевание |
Продолжительность кровотечения |
Время свертывания |
Активированное частичное тромбопластиновое время |
Протромбиновое время |
|
I |
Афибриногенемия |
Варьирует |
Свертывание не происходит |
Свертывание не происходит |
Свертывание не происходит |
|
II |
Гипопротромбинемия |
Нормальная |
Нормальное- продолжительное |
Варьирует |
Длительное |
|
V |
Парагемофилия |
Нормальная |
Продолжительное |
Продолжительное |
Продолжительное |
|
VII |
Недостаточность фактора VII |
Нормальная |
Нормальное |
Нормальное |
Продолжительное |
|
VIII |
Гемофилия А |
Нормальная |
Нормальное- продолжительное |
Продолжительное |
Нормальное |
|
VIII |
Болезнь Виллебранда |
Продолжительная |
Варьирует |
Варьирует |
Нормальное |
|
IX |
Болезнь Кристмаса, гемофилия В |
Нормальная |
Нормальное- продолжительное |
Продолжительное |
Нормальное |
|
X |
Недостаточность фактора Стюарта |
Нормальная |
Нормальное-продолжительное |
Продолжительное |
Продолжительное |
|
XI |
Недостаточность фактора XI |
Варьирует |
Нормальное- продолжительное |
Продолжительное |
Нормальное |
|
XII |
Симптом Хагемана |
Нормальная |
Продолжительное |
Продолжительное |
Нормальное |
|
XIII |
Недостаточность фактора, стабилизирующего фибрин |
Нормальная |
Нормальное |
Нормальное |
Нормальное |
|
Прекалликреин |
Симптом Флетчера |
Нормальная |
Продолжительное |
Продолжительное |
Нормальное |
|
Высокомолекулярный кининоген |
Симптом Фитцжеральда |
Нормальная |
Продолжительное |
Продолжительное |
Нормальное |
У пациентов с аутосомно-доминантой болезнью Виллебранда помимо недостаточности фактора VIII имеется нарушение в адгезии тромбоцитов. Между тем у больных гемофилией А отсутствует только свертывающая активность фактора VIII, при этом адгезия тромбоцитов не нарушена. Фактор адгезии тромбоцитов (фактор Виллебранда) синтезируется клетками эндотелия сосудов и мегакариоцитами (клетками-предшественниками тромбоцитов); он представляет собой крупный гликопротеин с мол. массой более 200000. Фактор Виллебранда обнаруживается в плазме и тромбоцитах в составе комплекса с молекулой фактора VIII. По-видимому, на поверхности тромбоцитов имеется рецептор гликопротеиновой природы, связывающий комплекс фактора Виллебранда с фактором VIII. Фактор Виллебранда, вероятно, стабилизирует прокоагулянтную активность фактора VIII. Болезнь Виллебранда может быть результатом наследуемого дефекта в олигосахаридном фрагменте гликопротеинового фактора Виллебранда. Аномальный олигосахарид может препятствовать адгезии тромбоцитов и дестабилизировать фактор VIII. При гемофилии А имеется дефект фактора VIII; при этом нарушается его свертывающая активность, в то же время адгезия тромбоцитов, определяемая фактором Виллебранда, не меняется. Фактор VIII представляет собой гликопротеин, содержащий 2300 аминокислот; его молекула обнаруживает частичную гомологию с церулоплазмином и фактором V. Синтезируется этот фактор в печени, селезенке и почках.
Тесты на свертывание крови
Для ознакомления с различными методиками, позволяющими оценить функционирование системы свертывания крови, рекомендуем читателю обратиться к соответствующим разделам учебников по физиологии или гематологии.
Антикоагулянты
Нормальная плазма характеризуется несколькими видами антитромбиновой активности. Небольшой вклад в нее вносит а1-антитрипсин. На долю специфического а2-глобулина приходится около 25% всей антитромбиновой активности плазмы. Он образует необратимый комплекс с тромбином и другими протеазами, препятствуя таким образом связыванию этих ферментов с их природными субстратами. а2-Глобулин рассматривается как а2-ингибитор плазмина, поскольку он инактивирует также плазмин, являющийся сериновой протеазой с фибринолитической активностью.
Наибольшая антитромбиновая активность присуща антитромбину III. Антитромбин III обладает незначительной эндогенной активностью и сильно активируется в присутствии гепарина, обладающего большим отрицательным зарядом. Гепарин, по-видимому, связывается со специфическим катионным участком антитромбина III, вызывая конформационное изменение его молекулы. В результате этого изменения антитромбин III приобретает возможность связываться со всеми сериновыми протеазами, включая трипсин, химотрипсин и плазмин. В системе свертывания крови антитромбин III ингибирует активность тромбина, факторов IХа, Ха, ХIа и ХIIа. У индивидов с наследственной недостаточностью антитромбина наблюдается склонность к образованию тромбов. Отсюда можно сделать вывод, что антитромбин выполняет физиологические функции и что в норме процесс свертывания крови у человека представляет собой очень динамичную систему.
Гепарин часто используется в клинической практике в качестве препарата, предотвращающего свертывание крови. Главным фактором, определяющим противосвертывающую активность гепарина, является активация им антитромбина III, который в свою очередь ингибирует рассмотренные выше сериновые протеазы. Известно, что небольшое количество гепарина находится на стенках сосудов, вследствие этого снижается активация внутреннего пути. Противосвертывающую активность гепарина можно подавить сильно катионными полипептидами (например, протамином). Такие полипептиды конкурируют с катионными участками антитромбина III за связывание с полианионным гепарином.
Препараты группы кумарина ингибируют витамин-К-зависимое карбоксилирование остатков Glu, приводящее к образованию Gla в N-концевой части молекулы факторов II, VII, IX и X. Все эти факторы синтезируются в печени, и образование остатков Gla необходимо для их созревания и, следовательно, для нормального функционирования внутреннего, внешнего и общего конечного путей свертывания. По-видимому, препараты кумарина ингибируют восстановление хиноновых производных витамина К в активные гидрохиноновые формы. Введение витамина К снимает блок, вызываемый кумарином, и обеспечивает созревание в печени Gla-зависимых факторов свертывания. Обращение действия кумарица витамином К наблюдается только через 12—24 ч; обращение же противосвертывающей активности гепарина протамином происходит практически сразу; это различие обусловлено природой антагонистических механизмов.
Фибринолиз
Имеются убедительные данные, свидетельствующие о том, что система свертывания крови в норме находится в динамическом равновесии, при котором фибриновые сгустки постоянно образуются, а затем растворяются. Плазмин представляет собой сериновую протеазу, способную гидролизовать фибриноген и фибрин, факторы V и VIII, факторы комплемента и различные полипептидные гормоны. В норме плазмин содержится в плазме в форме неактивного профермента (плазминогена). В большинстве тканей организма имеются активаторы плазминогена различных типов. Тканевый активатор плазминогена — это сериновая протеаза, каталитически неактивная в отсутствие контакта с фибрином. Находясь в контакте с фибрином, активатор плазминогена способен расщеплять молекулу плазминогена с образованием плазмина. Когда плазмин гидролизует фибрин, активатор плазминогена теряет свою активность и протеолиз затухает. Таким образом, обеспечивается эффективная регуляция процесса фибринолиза. Весьма перспективным представляется использование в терапевтических целях тканевого активатора плазминогена (ТАП), получаемого методами генной инженерии. ТАП способствует восстановлению проходимости коронарных артерий, снижая, таким образом, повреждение миокарда, происходящее при остром тромбозе коронарных сосудов. Еще один активатор плазминогена — протеолитический фермент урокиназа — содержится в моче. Урокиназа — это тоже сериновая протеиназа; она может активировать плазминоген, расщепляя его в двух местах.
Плазминоген в норме соосаждаeтся с фибрином и, следовательно, входит в состав фибринового сгустка. Образующийся в результате активации плазмин расщепляет молекулы фибрина на растворимые фрагменты, и сгусток исчезает (растворяется). Фибриновые сгустки с поперечными сшивками, труднее растворяются плазмином.
Концентрация активаторов плазминогена повышается при ряде заболеваний, в том числе при некоторых формах рака и при шоке. Антиплазминовая активность, обусловленная а1-антитрипсином и а2-ингибитором плазмина, может снижаться при циррозе печени. Некоторые бактериальные продукты. например стрептокиназа, способны активировать плазминоген без расщепления его молекулы и могут быть ответственны за диффузные кровоизлияния, наблюдаемые иногда у больных с диссеминированными бактериальными инфекциями.
Литература
Deykin D. Thrombogenesis, N. Engl. J. Med., 1967, 276, 622.
Genton E. et al. Platelet-inhibiting drugs in the prevention of clinical thrombotic disease. (2 parts), N. Engl. J. Med., 1975, 293, 1236, 1296.
George J. TV., Nurden A. T., Phillips D. R. Molecular defects in interactions of platelets with the vessel wall, N. Engl. J. Med., 1984, 311, 1084.
Gitschier J. et al. Characterization of the human factor VIІІ gene, Nature, 1984, 312, 326.
Heimark R. L. et al. Surface activation of blood coagulation, fibrinolysis and kinin formation, Nature, 1980. 286, 456.
Jackson C. M., Nemerson Y. Blood coagulation, Annu. Rev.
Biochem., 1980, 49, 767.
Kane W. H. et al. Factor Va-dependent binding of factor Xa to human platelets, J. Biol. Chem., 1980, 255, 1170.
McKee P. A. Hemostasis and disorders of blood coagulation. In: The Metabolic Basis of Inherited Disease, 5th ed., Stan- bury J. B. et al. (eds.), McGraw-Hill, 1983.
Stenflo J., Suttie J. W. Vitamin К-dependent formation of gamma-carboxyglutamic acid, Annu. Rev. Biochem., 1977, 46, 157.
Stites D. P., Stobo J. D., Wells J. V. Basic and Clinical Immunology, 6th ed., Appleton and Lange, 1987.
Weiss H. J. Platelet physiology and abnormalities of platelet function (2 parts), N. Engl. J. Med., 1975, 293, 531, 580.