БИОХИМИЯ - Л. Страйер - 1984
ТОМ 1
ЧАСТЬ I. КОНФОРМАЦИЯ И ДИНАМИКА
ГЛАВА 7. МЕХАНИЗМ ДЕЙСТВИЯ ФЕРМЕНТОВ: ЛИЗОЦИМ И КАРБОКСИПЕПТИДАЗА
7.4. Способ связывания конкурентного ингибитора
Рентгеноструктурное исследование комплекса лизоцима с три-NAG позволило установить локализацию активного центра и выявило, какие взаимодействия обеспечивают специфическое связывание субстрата; на этой основе была разработана гипотеза, предсказывающая детальный механизм ферментативного действия лизоцима. Оказалось, что три-NAG присоединяется к лизоциму в щели на его поверхности, занимая при этом примерно половину щели. Связывание происходит в результате образования водородных связей и вандерваальсовых взаимодействий. Электростатические взаимодействия отсутствуют, поскольку в три- NAG нет ионных групп.
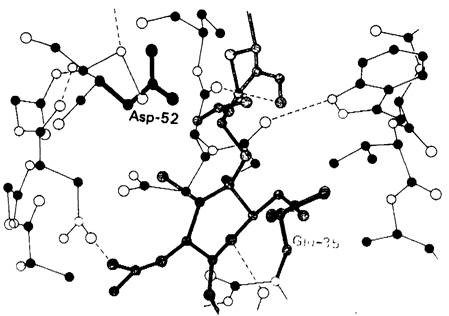
Водородные связи между три-NAG и лизоцимом показаны на рис. 7.9. Карбоксильная группа аспартата-101 образует водородные связи с остатками А и В. Наиболее специфические и прочные водородные связи образуются между ферментом и остатком С ингибитора. Здесь появляются четыре водородные связи. NH-группа индольного кольца триптофана-62 соединяется водородной связью с кислородом при С-6. Соседний аминокислотный остаток - триптофан-63 — аналогичным образом связывается с кислородом, стоящим при С-3. Когда три-NAG присоединяется к ферменту, кольцо триптофана-62 перемещается на 0,75 А. Прочные водородные связи формируются между СО- и NH-группами ацетамидной боковой цепи остатка сахара С и NH- и СО-группами основной цепи белка, принадлежащими соответственно 59-му и 107-му аминокислотному остатку.
Рис. 7.9. Водородные связи между три- NАG и лизоцимом. Участвующие в образовании водородных связей химические группы субстрата показаны синим, соответствующие группы фермента - красным
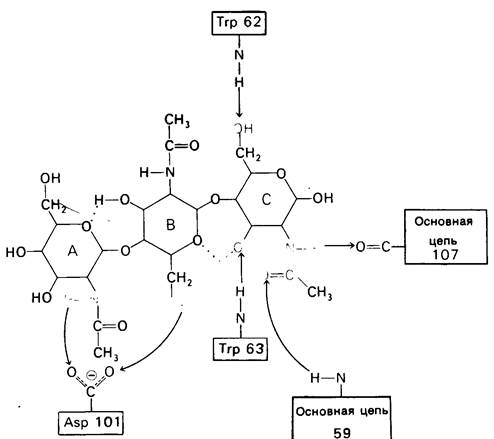
Между три-NAG и ферментом возникает большое число контактов, обусловленных вандерваальсовыми взаимодействиями. Остаток сахара В вовлечен в малое количество полярных контактов с ферментом, но он тесно связан с индольным кольцом триптофана-62. Остаток А довольно слабо контактирует с ферментом.
7.5. От структуры фермента-к механизму ферментативного действия
1. Как происходит связывание субстрата? Мы уже говорили о том, что метод рентгеноструктурного анализа не дает возможности непосредственно определить, как происходит связывание с ферментом эффективного субстрата. Однако данные, полученные при рентгеноструктурном анализе комплекса фермент—конкурентный ингибитор, могут сыграть ключевую роль в решении этой проблемы. Три-NAG заполняет только половину щели в молекуле лизоцима. Это очень многообещающее исходное положение. Было сделано допущение, что наблюдаемое связывание три-NAG как ингибитора происходит с образованием тех же связей, которые возникают и при связывании субстрата. Вполне вероятно, что для образования реакционноспособного ЕS- комплекса требуются дополнительные остатки сахара, способные заполнить вторую половину щели. Действительно, после присоединения три-NAG в щели остается место еще для трех остатков сахара. Это обнадеживало, поскольку было известно, что гексамер N-ацетилглюкозамина (гекса-NAG) быстро гидролизуется ферментом.
При тщательном построении моделей в щели на ферменте поместились три дополнительных остатка сахара, обозначенных D, Е и F (рис. 7.10). Остатки Е и F подошли прекрасно, образуя несколько прочных водородных связей и контактов, обусловленных вандерваальсовыми взаимодействиями. Однако остаток D входил в щель только при условии некоторой деформации. При нормальной конформации (в виде кресла) его атомы С-6 и O-6 оказывались слишком сближенными с некоторыми группами на ферменте.
Рис. 7.10. Способ связывания гекса-NAG (показан желтым) с лизоцимом. Расположение углеводных остатков А, В и С (слева) соответствует локализации три-NAG в комплексе с лизоцимом; расположение остатков D, Е и F (справа) предсказали путем модельного построения. Зеленым показаны два аминокислотных остатка, непосредственно участвующих в катализе
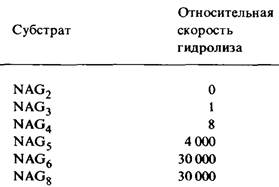
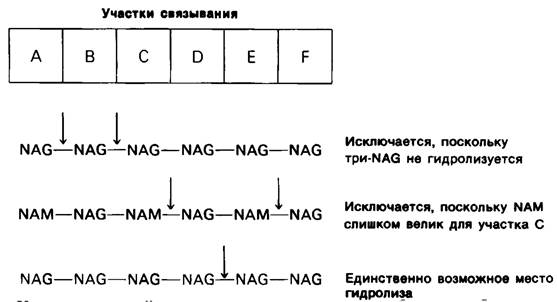
2. Какая из связей расщепляется ферментом? Скорость гидролиза олигомеров N- ацетилглюкозамина стремительно возрастает при увеличении числа остатков сахара от 4 до 5, т.е. от NAG4 до NAG5 (табл. 7.1) Удлинение субстрата еще на один остаток (NAG6) дает дополнительное увеличение скорости расщепления; однако возрастание числа остатков сахара в субстрате до 8 уже не оказывает действия. Эти данные согласуются с результатами рентгеноструктурного исследования, показавшими, что имение шести остатков сахара достаточно для заполнения щели, где расположен активный центр.
Таблица 7.1. Эффективность олигомеров N-ацетилглюкозамина в качестве субстратов
Какая из связей в гекса-NAG расщепляется ферментом? Исходя из того, что три NAG не подвергается расщеплению, можнo считать, что связь А—В (т.е. гликозидная связь между остатками А и В)-это не та связь, на которую действует фермент. Аналогично этому фермент не может расщеплять и связи В—С. Второе и решающее доказательство того, что связь В—С не расщепляется ферментом, состоит в том, что NAM не может встать в положение остатка С. Если NAG отлично умещается в участке С, то NAM здесь не умещается из- за лактильной боковой цепи. Между тем в полисахариде клеточной стенки бактерий лизоцим расщепляет связь NAM—NAG. Следовательно, и связь С—D не может подвергаться расщеплению, если действительно полисахарид клеточных стенок бактерий связывается с лизоцимом таким же образом, как и гекса-NAG. Несоответствие NAM положению С исключает еще одно место гидролиза, а именно связь Е—F. Вспомним, что полисахарид клеточной стенки-это чередующийся полимер NAM и NAG; следовательно, если NAM не может занимать положение С, то этот остаток не может стоять и в положении Е.
Из приведенных соображений вытекает, что при расщеплении ферментом гексамерного субстрата связи А—В, В—С, С—D и Е—F не могут разрываться. Следовательно, единственным возможным местом расщепления субстрата является связь D—Е (рис. 7.11).
Рис. 7.11. Ход рассуждений, доказывающих, что место приложения действия фермента - гликозидная связь между углеводными остатками D и Е
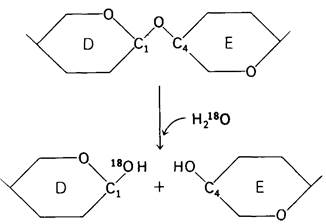
3. Какая группа на ферменте непосредственно осуществляет катализ? Заключение о том, что гидролиз субстрата происходит по связи D—Е, позволило перейти далее к выявлению тех групп на ферменте, которые непосредственно осуществляют реакцию гидролиза. Для этого, однако, нужно еще более точно локализовать место расщепления субстрата, а именно выяснить, по какую сторону от гликозидного атома кислорода происходит разрыв связи. Ответ на этот вопрос был получен в опытах по ферментативному гидролизу в среде, содержащей воду, меченную стабильным тяжелым атомом кислорода 18O (рис. 7.12). В выделенных по окончании гидролиза сахарах 18O оказался присоединенным к С-1 остатка Dтогда как гидроксильная группа при С-4 в остатке Е содержала обычный изотоп кислорода. Отсюда следует, что при гидролизе разрыв связи происходит между С-1 остатка О и кислородом гликозидной связи, примыкающим к остатку Е. Эта работа может служить примером использования изотопов в изучении механизма ферментативного катализа. Без изотопов было бы крайне трудно, а может быть, и невозможно в данном случае установить точное место приложения действия фермента.
Рис. 7.12. Проведение гидролиза в меченной 18O воде показало, что лизоцим расщепляет связь между С-1 и О, но не связь между С-4 и О (изображен только скелет остатков D и Е)
Затем перешли к поиску возможных каталитических групп, которые должны располагаться вблизи расщепляемой гликозидной связи. Как указывалось, в предыдущей главе, под каталитическими группами подразумевают те группы фермента, которые непосредственно участвуют в образовании или разрыве ковалентных связей. Наиболее подходящие кандидаты на эту роль, это группы, способные к образованию водородных связей в качестве доноров ши акцепторов водорода. Отрыв или присоединение иона водорода - это критический этап большинства ферментативных реакций. В лизоциме единственные остатки, способные быть каталитическими и расположенные вблизи расщепляемой гликозидной связи, это аспартат-52 и глутамат-55. Остаток аспарагиновой кислоты лежит по одну сторону от гликозидной связи, а остаток глутаминовой-по другую. Окружение этих двух кислотных боковых цепей совершенно различно. Аспартат-52 находится в полярном окружении, где он служит акцептором водорода в сложной сети водородных связей. Глутамат-35, напротив, расположен в неполярной области. Отсюда следует, что при pH 5, оптимальном pH для гидролиза лизоцимом хитина, аспарагиновая кислота в положении 52 должна находиться в ионизированной СOO -форме, тогда как глутаминовая кислота в положении 35-в неионизированной СООН-форме. Расстояние между гликозидной связью и ближайшим к ней атомом кислорода как одной, так и другой кислотной группы составляет примерно 3 А (рис. 7.13).
Рис. 7.13. Структура части активного цен грализоцима. Желтым показаны кольца D и Е субстрата гекса-NAG. Вблизи субстрата расположены боковые цепи аспартата-52 (красное) и глутамата-35 (зеленое)