Структура и функционирование белков. Применение методов биоинформатики - Джон Ригден 2014
Интегральные серверы для предсказания функции по структуре
Введение
Методы предсказания структура-функция
Как было показано в предыдущих главах, есть очень большое количество различных методов предсказания функции белка на основе его структуры. Их описание и рассмотрение пригодности приведены в нескольких обзорах (Kim et al. 2003; Watson et al. 2005; Rigden 2006). Ни один из методов не является совершенным и ни от одного из них нельзя ожидать успешного применения во всех случаях. Например, некоторые методы подходят только для ферментов и совершенно не в силах помочь, если рассматриваемый белок таковым не является. Другие методы сильно полагаются на некое совпадение, - будь то укладка, или мотив, или сайт связывания и так далее, - с белком, чья структура известна. Поэтому если такое совпадение найти нельзя, или оно оказывается совпадением с другим гипотетическим белком, то эффективность такого метода обращается в ноль.
Как следствие, разумный подход состоит в том, чтобы направить большое число этих методов на структуру белка и посмотреть, что выпадет. Именно так и поступают два сервера, описываемые в этой главе. Это ProKnow из Университета Калифорнии (UCLA) (http://proknow.mbi.ucla.edu) и ProFunc из Европейского института биоинформатики (European Bioinformatics Institute, ЕВІ) (http://www.ebi.ac.uk/profunc). Оба они используют предсказания, основанные как на последовательности белка, так и на его структуре, и широко автоматизированы: загружаешь файл в формате PDB и терпеливо дожидаешься результатов.
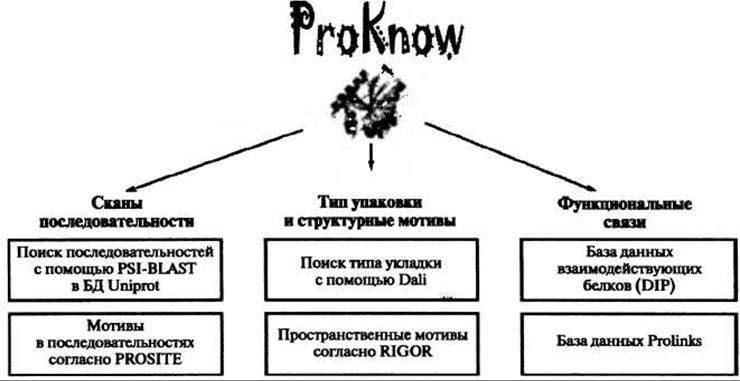
Рис. 10.1. Схематическая диаграмма основанных на структуре и последовательности методов, применяемых к пространственной структуре любого белка, загруженной на сервер предсказания функции ProKnow. Основанные на последовательности методы - это PSI-BLAST (Altschul et al. 1997) и PROSITE (Hulo et al. 2004). Основанные на структуре методы - это поиск по фонду Dali (Holm and Sander 1998) и поиск по пространственным мотивам RIGOR (Kleywegt 1999). Последние два метода для определения интересных функциональных связей для лучших результатов программы PSI-BLAST используют результаты базы данных взаимодействующих белков DIP (Database of Interacting Proteins, Xenarios et al. 2002) и базы данных Prolinks (Bowers et al. 2004). Обобщая все результаты, получают функциональные аннотации генной онтологии (GO) и комбинируют их, используя байесовское присвоение весов, для получения набора предсказаний функции и соответствующих оценок надежности
Для иллюстрации двух этих методов в действии в качестве примера мы взяли только что полученную пространственную структуру. Это структура предполагаемой ацетилтрансферазы из Vibrio cholera, полученная в 2005 году в Центре по структурной геномике на Среднем Западе (MCSG). Она была опубликована в базе данных PDB 28 февраля 2006 года с кодом 2fck (Cuff et al. 2007). На момент появления структуры функция белка была известна только предварительно; последовательность имела более 50% идентичности с серин-ацетилтрансферазой рибосомальных белков (ribosomal-protein-serine acetyltransferase) и содержала несколько мотивов, характерных для ацетил- трансферазной активности. Раз структура была известна, эти предварительные описания функции получили мощную поддержку, поскольку оказалось, что есть сильное структурное сходство, как глобальное, так и локальное, с другими - удаленными - ацетилтрансферазами. Наиболее сильные сходства были обнаружены в вероятном сайте связывания, где мог бы связаться коэн- зим А (соА). Некоторые из этих сходств будут рассмотрены ниже.