Структура и функционирование белков. Применение методов биоинформатики - Джон Ригден 2014
Распознавание фолда
Введение
Предсказание структуры ab initio и моделирование по гомологии
Если мы надеемся однажды описать структуру сколь-нибудь заметной доли белков в природе, не прибегая к открытию каких-либо революционных экспериментальных методов, то нам понадобится подход, который позволит предсказывать структуру на основе последовательности с использованием вычислительных методов. После того, как в 1961 г. Анфинсен показал, что после денатурации рибонуклеаза может проходить процесс рефолдинга, сохраняя при этом ферментативную активность, стала популярной идея о том, что информация, необходимая белку для обретения окончательной конформации, закодирована в его последовательности. В результате для прогнозирования структуры белков течение последних десятилетий использовалось сочетание “чистых” методов, в которых в качестве входных данных используется лишь аминокислотная последовательность, и законов физики (или их приближений). В этом направлении были достигнуты определенные успехи, которые описаны в главе 1 настоящей книги. Однако в целом эти методы отличаются либо громоздкими вычислениями, что делает их применение на практике затруднительным, либо низкой производительностью и неточными результатами при исследовании любых систем, кроме белков небольшого размера (менее 100 аминокислотных остатков). Физический подход может показаться единственно верным решением проблемы сворачивания, однако предсказание структуры белков имеет большое практическое значение, а значит, необходимо принять имеющиеся ограничения и двигаться, хотя бы и временно, в направлении поиска более прагматичного решения. Такой подход привел к тому, в области методов предсказания белковой структуры акцент сместился от физики к глубинному анализу, или “добыче”, данных (data mining).
Давно известно, что похожие белковые последовательности сворачиваются в похожие структуры. Потому, имея новую белковую последовательность, структуру которой предстоит установить, (называемую в дальнейшем “исследуемая последовательность”) достаточно просто проверить, существуют ли похожие последовательности, структура которых уже известна. Если существуют последовательности с высокой степенью подобия, процесс определения структуры легко осуществить, применяя методы выравнивания аминокислотных последовательностей. Используя простой способ оценки подобия типов аминокислот, такой как оценочная матрица BLOSUM, в сочетании с алгоритмом динамического программирования, таким как алгоритм Смита-Уотермана, можно быстро и оптимально (согласно оценочной функции) выровнять две последовательности.
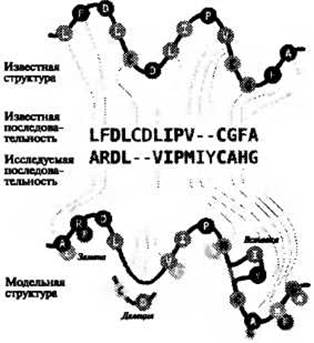
Рис. 2.1. (Цветную версию рисунка см. на вклейке.) Схематичное представление упрощенного алгоритма построения модели с помощью выравнивания последовательностей исследуемого белка и шаблона. Показано выравнивание последовательности известной структуры (“известная последовательность”) и исследуемой последовательности. Размытыми линиями показаны вставки и делеции; красными буквами - аминокислотные замены. Остатки окрашены согласно биофизическим свойствам. Тонкие волнистые линии соединяют соответствующие положения в исследуемой и известной последовательности.
Имея выравнивание последовательности по известной структуре (далее называемой “шаблон”), можно затем построить грубую модель простым копированием соответствующих пространственных координат шаблона и переименованием аминокислотных остатков в соответствии с эквивалентными остатками из выравнивания (рис. 2.1).
Далее модель можно улучшить, используя множество методов моделирования по гомологии, описанных в соответствующей главе настоящей книги. Преимущества этого подхода очевидны: он является быстрым в вычислительном отношении, а точность итоговой модели очень высока при условии высокой степени сходства исследуемой последовательности и последовательности шаблона. Это обстоятельство немедленно указывает на ограничение метода. Если нет похожей последовательности с уже известной структурой, то никакого результата не удастся получить вовсе.
Таким образом, поиск решения для проблемы предсказания белковых структур осуществляется по двум направлениям. Первое направление, основанное на общих физических принципах, нацелено на создание понятного и универсального метода, который позволял бы предсказывать структуру по последовательности, а также предоставлял бы возможности для дизайна белков, изучения динамики и решения множества других важных задач. Однако создание такого метода сопряжено с экспериментальными сложностями и, вероятно, в ближайшие годы останется нереализованным с вычислительной точки зрения. Другое направление представляет собой простой и понятный, но существенно ограниченный эвристический метод моделирования по гомологии, с помощью которого можно получать модели высокого качества, но лишь в очень ограниченном числе случаев. Именно для решения этой проблемы существует метод, известный как “распознавание фолда”, который был создан, чтобы соединить два противоположных направления исследований.