ХИРУРГИЯ НОВОРОЖДЕННЫХ - 1976
2. ЧАСТНЫЕ РАЗДЕЛЫ
8. Пороки развития отдельных органов и систем
Пороки развития пищевода
Среди пороков развития пищевода наиболее часто встречаются атрезии, трахеопищеводные соустья, частичные стенозы, халазия, ахалазия. Дети с пороками развития пищевода, как правило, нуждаются в неотложном оперативном лечении. Атрезия пищевода, по данным Г. А. Баирова (1968), наблюдается у одного из 3500 новорожденных. В Финляндии один ребенок из 3000 рождается с атрезией пищевода (Louhimo, 1970). Эмбриогенез пищевода и основные пороки его развития схематически изображены на рис. 46.
Рис. 46. Формирование пищевода, возможные механизмы развития пороков и их основные варианты.
Пищевод и трахея закладываются в начальных отделах общей первичной кишечной трубки. Зона будущего пищевода отмечена красным цветом (а). Далее посередине дна глотки появляется желобок, который быстро превращается в трубчатый вырост — трахею (б), проходящую параллельно кишечной трубке. Каудальный конец трахеи начинает увеличиваться и раздваиваться (в), образуя закладки легких. Дистальнее глотки пищеварительная трубка заметно суживается, образуя пищевод (б, в, г). Направления нормального роста и разделения указанных образований показаны на рис. в стрелками.
При несоответствии направления и скорости роста трахеи и пищевода (рис. д сравнить с рис. в) возможно развитие атрезии пищевода с трахеопищеводным свищом (Gruenwatd).
Вероятной причиной развития атрезии и стенозов пищевода, а также всего пищеварительного тракта считают нарушение процессов вакуолизации в солидной стадии, которую пищевод проходит вместе с другими образованиями кишечной трубки в сроки от 20-го до 40-го дня. На рис. е показаны возможные исходы разрастания эпителия при разрешении солидной стадии — нормальное восстановление просвета пищевода, атрезия, стеноз. На рис. ж схематически представлены основные клинические варианты атрезии пищевода.
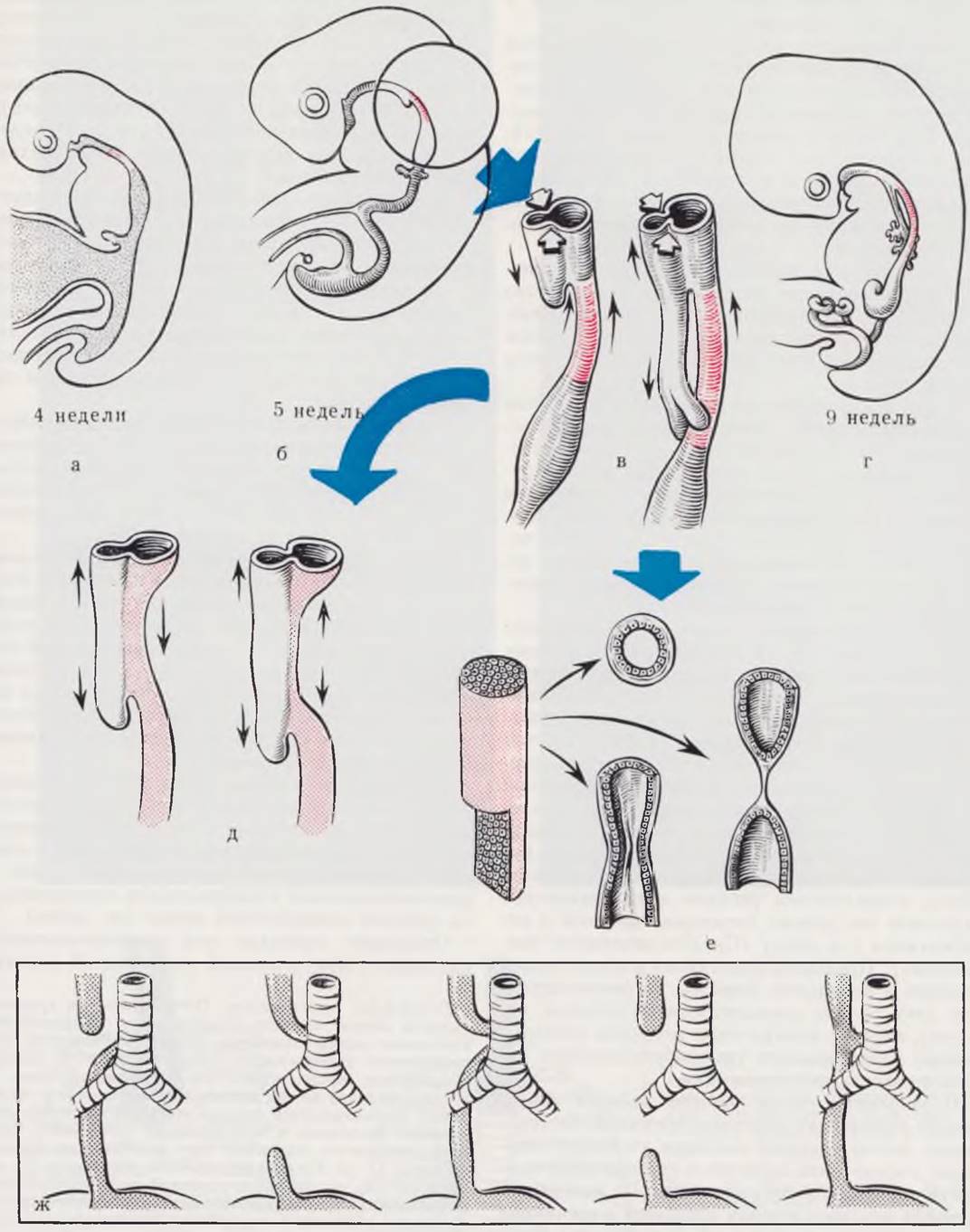
Самым частым пороком развития пищевода, который имеет место у 85 — 95% всех больных (Guendet, 1968; Кummern, 1970; Коор, 1971), является атрезия верхнего приводящего отрезка с образованием нижнего трахеопищеводного свища.
Клиническая картина. Проявления атрезии пищевода отчетливы. Важно помнить о возможности наличия этого порока развития. Уже через 2 — 3 ч после рождения верхний слепой отрезок пищевода и носоглотки переполняются слизью, у ребенка появляются обильные пенистые выделения изо рта. Часть слизи аспирируется, возникают приступы цианоза. На первые появления пенистых выделений персонал родильного дома, как правило, не обращает должного внимания, сестра отсасывает содержимое носоглотки, и состояние ребенка улучшается, но ненадолго. Пенистые выделения изо рта и приступы цианоза упорно повторяются, быстро развиваются признаки аспирационной пневмонии.
Такие упорные пенистые выделения изо рта и приступы цианоза дают возможность заподозрить неблагополучие со стороны пищевода уже в первые 4 — 5 ч после рождения ребенка, во всяком случае до первого кормления. Диагноз уточняют путем катетеризации пищевода тонким уретральным катетером с закругленным концом. Катетер через нос вводят в слепой отрезок пищевода, шприцем отсасывают слизь. При последующем введении воздуха через катетер он с шумом выделяется из носоглотки (положительный симптом Элефанта). Если верхний отрезок пищевода сообщается с трахеей, в клинической картине будут превалировать нарушения дыхания, так как содержимое верхнего отрезка пищевода попадает через свищ в трахею.
В последние годы большинство зарубежных авторов (Romstadt, 1966; Raffensperger, 1970; Velona, 1971) настойчиво рекомендуют зондировать пищевод всем без исключения новорожденным. Обязательное зондирование пищевода введено в Киеве, Риге, Ленинграде. Этим простым приемом без труда диагностируют пороки развития пищевода тотчас после рождения, что дает возможность своевременно направить ребенка в хирургическое отделение.
Наличие нижнего трахеопищеводного соустья диагностируют при осмотре и перкуссии брюшной полости. При отсутствии сообщения нижнего отрезка пищевода с трахеей воздух не попадает в желудок и кишечник, живот запавший, при перкуссии над всеми отделами брюшной полости определяется тупой звук. У больных с нижним трахеопищеводным свищом желудок и кишечник раздуты воздухом, что легко выявляется при осмотре и перкуссии живота.
Таким образом, диагностика и даже определение формы атрезии пищевода у большинства больных не представляют затруднений. Даже без катетеризации пищевода диагноз атрезии пищевода может быть поставлен в первые часы жизни ребенка.
Большие трудности представляет диагностика трахеопищеводных свищей без атрезии. В клинической картине у таких больных в зависимости от ширины соустья преобладают либо нарушения дыхания, особенно во время кормления ребенка в положении на левом боку, либо рецидивирующие пневмонии. Диагноз в этих случаях уточняют методом рентгенологического исследования или путем непосредственного осмотра свища при трахеобронхоскопии.
После установления диагноза исключают кормление через рот. В верхний слепой отрезок пищевода через нос вставляют тонкий, лучше двухпросветный, катетер, через который каждые 20 — 30 мин осторожно отсасывают слизь. Ребенку придают возвышенное положение, чем предупреждают заброс в легкие кислого содержимого желудка и развитие аспирационной пневмонии. В таком положении ребенка транспортируют в хирургическое отделение. Постоянное отсасывание слизи из верхнего отрезка пищевода во время транспортировки обязательно. Перед отправлением ребенка в хирургическое отделение ему делают инъекцию викасола и антибиотиков (см. главу «Предоперационная подготовка»). Проводить рентгенологическое исследование в родильном доме мы не рекомендуем. Это дает только дополнительную лучевую нагрузку, нередко контрастные вещества забрасываются из пищевода в трахеобронхиальное дерево и развивается пневмония.
В отделении после осмотра ребенка обязательно производят рентгенологическое исследование, целью которого является уточнение диагноза, определение наличия и степени выраженности аспирационной пневмонии. От контрастирования верхнего отрезка пищевода в последние годы клиника отказалась. У большинства больных для уточнения диагноза вполне достаточно рентгенограммы с рентгеноконтрастным катетером, введенным в проксимальный отрезок пище вода (рис. 47). Если возникает необходимость контрастирования пищевода, применяют йодолинол (не более 1 — 1,5 мл). После производства рентгенограммы йодолинол отсасывают и только после этого удаляют катетер. Оставление контрастного вещества в пищеводе (рис. 48) ведет к последующему забрасыванию его в трахеобронхиальное дерево.
Рис. 47. Рентгенограмма грудной клетки новорожденного. Диагноз: атрезия пищевода. Катетер контурирует верхний отрезок пищевода. Сообщения нижнего отрезка с трахеей нет.
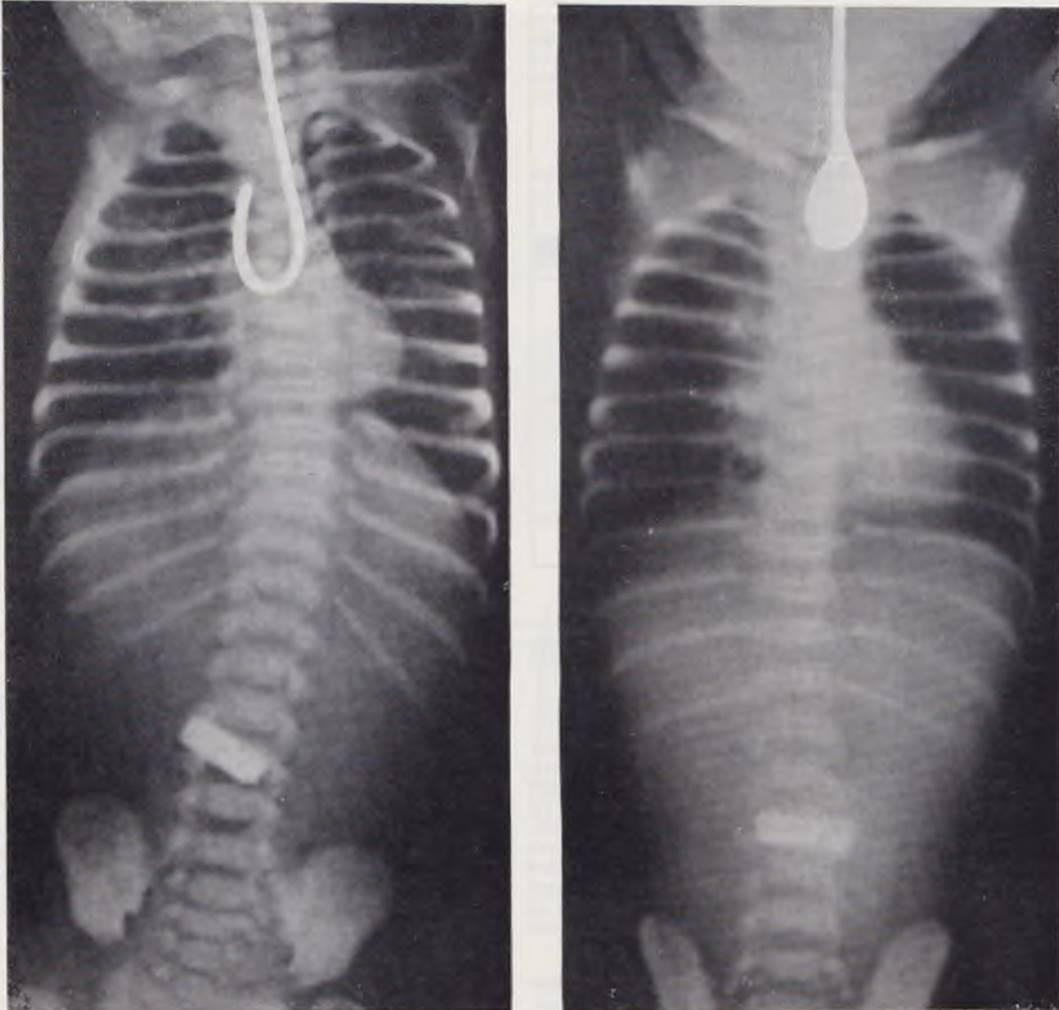
Рис. 48. Рентгенограмма грудной клетки новорожденного. Диагноз: атрезия пищевода. Оставленный в пищеводе йодолинол аспирирован в трахею. Массивная правосторонняя пневмония.
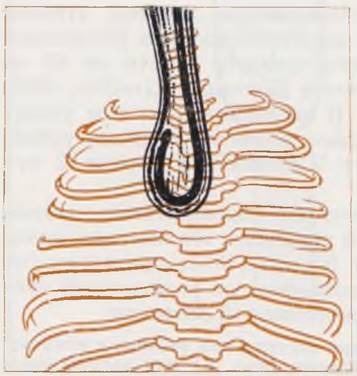
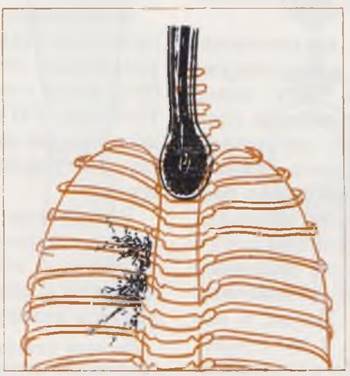
При трактовке изменений в легких следует помнить, что современные диагностические рентгеновские аппараты в большинстве случаев специально не приспособлены для исследования новорожденных. Получаемые на них рентгенограммы легких жесткие, что необходимо учитывать при чтении рентгенограмм. Действительная степень поражения легких у таких больных значительно больше, чем это определяется па рентгенограмме.
У детей с атрезией пищевода нередки сопутствующие пороки развития, которые, по данным разных авторов, встречаются в 20 — 50% случаев (Г. А. Баиров, 1968; Okmian, 1966; Forrester, Cohén, 1970; Toyma, 1971), что значительно ухудшает результаты лечения.
Лечение. Лечение атрезии пищевода оперативное. Дети, поступившие в клинику в первые 10 — 12 ч после рождения, не требуют специальной предоперационной подготовки. При позднем поступлении, с явлениями выраженной аспирационной пневмонии, предоперационная подготовка и лечение пневмонии проводятся по общим правилам до улучшения состояния больного. Для обеспечения полноценного питания накладывают гастростому.
Большинство авторов применяют правосторонний экстраплевральный пли трансплевральный доступ. В последние годы при оперативном лечении атрезии пищевода методом выбора считают правостороннюю боковую торакотомию с рассечением кожи в вертикальном направлении по средней подмышечной линии (см. выше).
Операцию проводят под эндотрахеальным наркозом с искусственной вентиляцией легких.
Техника операции. После вскрытия грудной полости легкое отводят в медиальном направлении. Рассекают медиастинальную плевру, мобилизуют, перевязывают и пересекают непарную вену. В тканях средостения, ориентируясь на блуждающий нерв и трахею, находят концы пищевода. Нахождение и выделение проксимального отрезка облегчаются предварительным введением в него катетера. Наложение прямого анастомоза возможно при диастазе, не превышающем 1,5 см. Устранение диастаза в пределах 1,5 см проводят за счет преимущественной мобилизации проксимального отрезка. Мобилизация дистального отрезка может привести к нарушению кровоснабжения его и последующему расхождению анастомоза. После мобилизации пищевода дистальный отрезок его у места перехода в трахею прошивают капроновой лигатурой, перевязывают и отсекают. Методом выбора мы считаем наложение анастомоза конец в конец одним рядом швов на атравматической игле (№ 00000 — 000000) узлами внутрь по всей окружности анастомоза (рис. 49). При хорошо выраженной стенке дистального отрезка возможно применение двух рядов швов по Haight. При этом следует помнить, что малейшее натяжение ведет к прорезыванию, как правило, второго ряда швов на стенке дистального отрезка. От сложных швов полностью отказались.
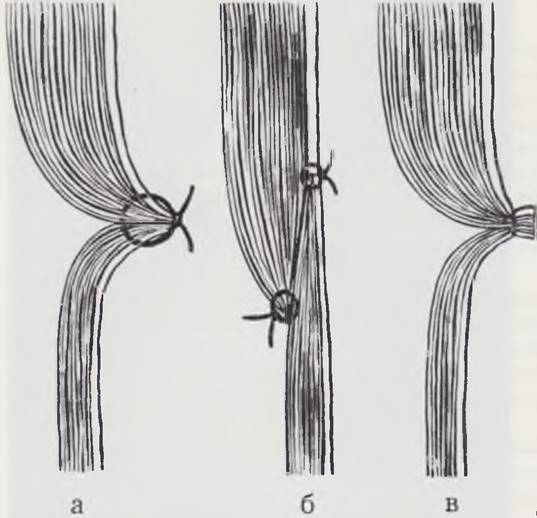
Рис. 49. Варианты пищеводного шва.
а — однорядный шов узлами внутрь по всей длине анастомоза; б — двухрядный телескопический анастомоз по Haight; в — механический шов.
В последние годы в хирургии пищевода у новорожденных находят применение механические швы с помощью различных сшивающих аппаратов (Г. А. Баиров, 1973; Ю. Л. Дорошевский, 1973; Okmian е. а., 1969).
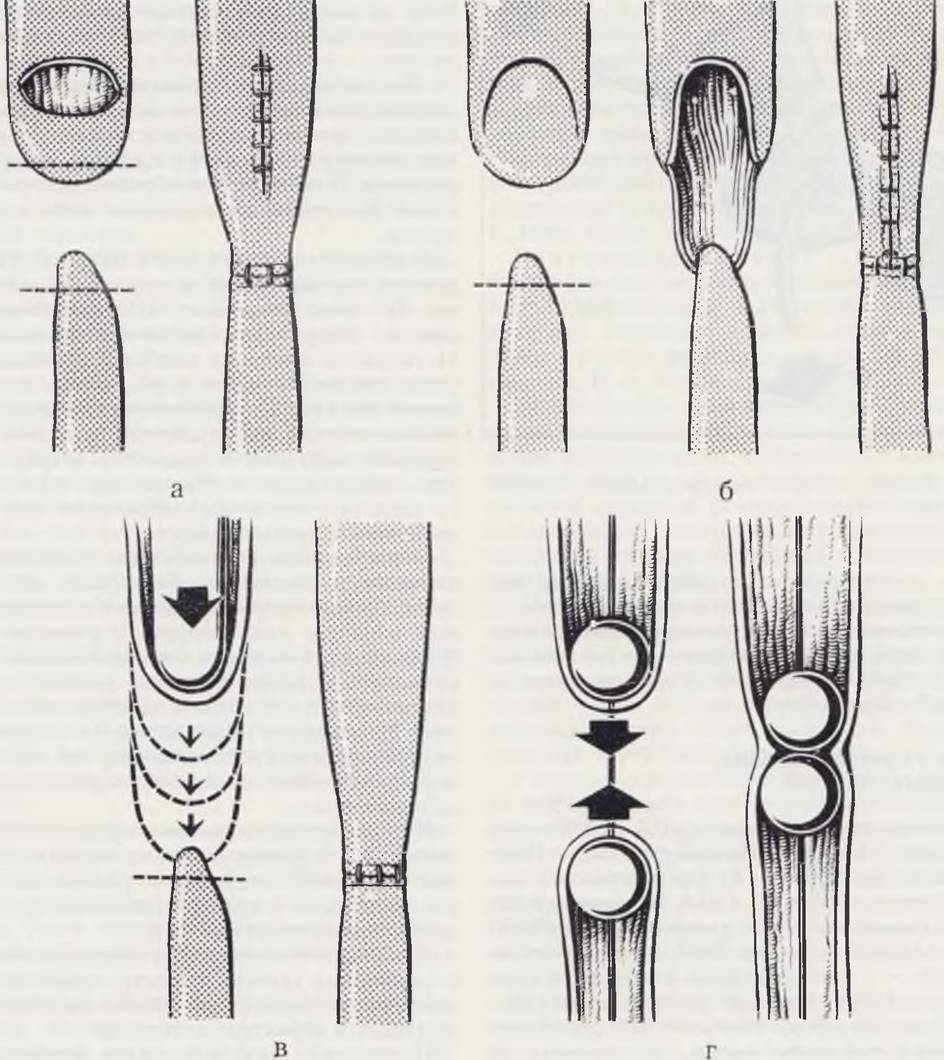
Sulamaa предлагает прошивать трахеопищеводное соустье без отсечения его и с последующим наложением анастомоза по типу конец проксимального в бок дистального отрезка применяет у отдельных больных при отсутствии диастаза и малом диаметре дистального отрезка. Удлинение пищевода на 4 — 5 мм возможно различными методами (рис. 50). Okmian проводит поперечное пересечение стенки проксимального отрезка пищевода через все слои на половину его окружности с последующим ушиванием рапы в продольном направлении. Метод, с нашей точки зрения, наиболее прост, и мы с успехом пользуемся им. Переднюю губу анастомоза,
как правило, ушивают на катетере с наружным диаметром не более 2 мм, который оставляют в пищеводе на 2 — 5 дней. Через катетер ребенка кормят, при необходимости отсасывают содержимое желудка.
Рис. 50. Варианты удлинения пищевода: по Оkmian (а); по Ten-Kate — Баирову (б); бужированием по Hovard (в); тракционное удлинение по Rehbein (г).
Попытка замещения дефекта пищевода из поперечной ободочной кишки по методу Уотерстона у новорожденных оправдана лишь при большом опыте хирурга, в совершенстве владеющего техникой эзофагопластики. Soave (1972) успешно произвел две такие операции. Он приводит сборные данные Holder (1963), где на 747 атрезий пищевода расхождения прямого анастомоза составили 16,7%. Для эзофагопластики при атрезии пищевода некоторые авторы (Vidue, Levy, 1970) применяют перикард, другие (Д. Е. Бабляк, 1973) предлагают выкраивать из него полоски, фиксируя ими пищевод выше и ниже линии швов анастомоза с целью ослабления натяжения последнего. Эти методы следует считать мало приемлемыми, ибо при несостоятельности швов анастомоза и последующем развитии гнойных внутриплевральных осложнений подвергается риску инфицирования открытая околосердечная сорочка.
После наложения анастомоза проводят контроль гемостаза. Восстанавливают непрерывность медиастинальной плевры. В сомнительных случаях плевральную полость дренируют на 2 — 3 дня. Рану послойно ушивают наглухо (дренаж выводят через отдельный прокол грудной стенки).
При невозможности наложения анастомоза операцию заканчивают прошиванием и перевязкой трахеопищеводного свища, гастро- и эзофагостомией. В более позднем возрасте производят пластику пищевода сегментом толстой кишки.
В послеоперационном периоде при отсутствии осложнений питание начинают через катетер, оставленный в пищеводе, уже на 2-е сут после операции. Плевральный дренаж удаляют на 2 — 3-й день.
Результаты оперативного лечения атрезии пищевода стали значительно лучше. По данным различных авторов, удается спасти от 43 до 85% детей с атрезией пищевода (Livaditis, 1968; Rickham, 1971). В последние годы нам удавалось спасти каждого второго — третьего ребенка, поступившего в клинику в первые 2 сут жизни.
Результаты лечения недоношенных, родившихся с атрезией пищевода, значительно хуже. В связи с этим Hovard (1965) предлагает отказаться у недоношенных от радикальной операции в пользу многоэтапной операции.
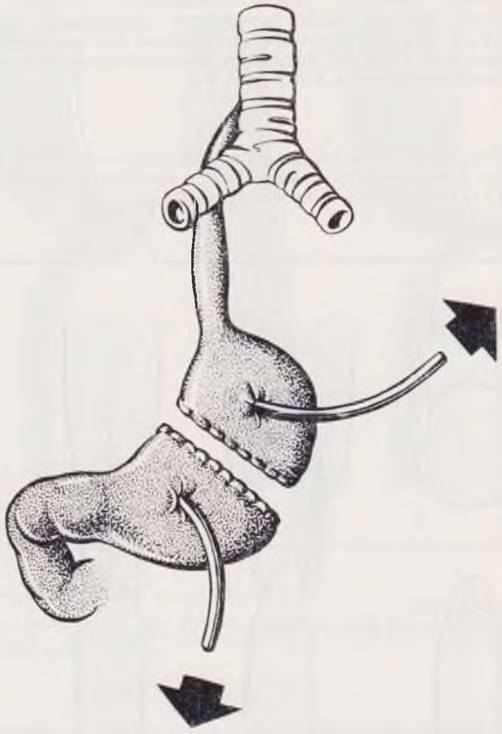
Первым этапом при поступлении ребенка в клинику производят гастростомию (рис. 51) или лапаротомию с последующим рассечением желудка на границе верхней и средней третей и выведением обоих концов его на брюшную стенку. В двенадцатиперстную кишку вводят зонд и налаживают энтеральное питание. Ребенок постоянно находится в положении с возвышенным головным концом. Содержимое проксимального слепого отрезка пищевода постоянно отсасывают через зонд. В проксимальный отрезок 2 — 3 раза в день вводят толстый эластичный резиновый зонд и производят осторожное растягивание пищевода. Таким образом, за 2 — 3 нед удается растянуть пищевод на 2 — 2.5 см. Следующим этапом производят торакотомию, разобщают трахео пищеводный свищ, анастомозируют концы пищевода. После заживления анастомоза и раны грудной стенки устраняют гастростому. Таким путем удается улучшить результаты лечения недоношенных детей. Об успешном применении этого метода сообщают Lafer и Boley (1966).
Рис. 51. Двойная гастростома с разделением желудка по Meecker.
Дети, оперированные по поводу атрезии пищевода, нуждаются в последующем диспансерном наблюдении с целью своевременного выявления и устранения возможного сужения пищевода (которое развивается после операции у каждого 3 — 5-го ребенка).
Пороки развития сердца и крупных сосудов
Пороки сердца встречаются у 0,3 — 1,3% новорожденных (Л. М. Болховитинова, 1961; В. В. Бадмаева, 1962; Б. А. Константинов и др., 1966; Ñamara, 1963). В США от врожденных пороков сердца ежегодно умирает около 10000 детей, в Англии — свыше 6000, причем больше 2/з из них — в первые недели и месяцы жизни (Aberdeen, 1969). Согласно нашим наблюдениям, на 1-м году жизни погибает 70% детей, родившихся с пороками сердца, из которых 1/3 умирает в первые 4 нед жизни. Среди новорожденных смертность от пороков сердца составляет от 9 до 11% общей смертности. М. Л. Петров-Маслаков и И. И. Климец (1965) утверждают, что на 1-й неделе жизни пороки сердца как причина неонатальной смертности уступают только родовой травме. Lambert (1966) считает их основной причиной летальности в этой возрастной группе. Между тем более половины детей, погибших в первые недели и месяцы жизни, имеют технически устранимые пороки (В. И. Бураковский, Б. А. Константинов, 1970).
Пороки сердца у новорожденных, хотя и составляют доминирующий вид поражения сердечно-сосудистой системы в этом возрасте (80 — 90% от числа всех органических заболеваний сердца), не исчерпывают всей патологии этой системы.
Все заболевания сердечно-сосудистой системы новорожденных делят на первичные, при которых имеется непосредственное повреждение сердца, и вторичные, при которых нарушения гемодинамики обусловлены расстройством функции или анатомии любого другого органа.
К первичным поражениям относят: 1) врожденные пороки сердца и сосудов; 2) миокардиты; 3) миокардиопатии; 4) артериовенозные свищи. Вторичные заболевания включают: 1) гиалиноз легочных мембран; 2) недостаточность кровообращения в результате гемолитической анемии; 3) кардиомегалию с недостаточностью атриовентрикулярных клапанов в результате асфиксии в родах; 4) острое «легочное» сердце после аспирационных осложнений; 5) недостаточность кровообращения при черепно-мозговой травме в родах.
При большом разнообразии этиологии и патогенеза названных заболеваний они имеют одну общую черту — чаще всего носят врожденный характер или связаны с родовым актом. К врожденной патологии относят пороки сердца и сосудов, которые являются результатом эмбриопатии или следствием хромосомных аберраций. Миокардиты новорожденных также имеют внутриутробное происхождение, так как многие вирусы способны проникать через плаценту от матери к плоду.
К поражениям сердечно-сосудистой системы, связанным с родовым актом, относят последствия асфиксии, аспирации и реанимации — острое «легочное» сердце, кардиомегалию при черепно-мозговой травме и др.
Особенности анатомии и гемодинамики у новорожденных таковы, что затрудняют не только топическую диагностику порока, но и определение общего характера патологии.
Клиническая картина различных пороков сердца отличается стереотипностью и преобладанием общих симптомов. Локальные признаки не успевают сформироваться и выражены скудно. Такой классический симптом порока сердца, как шум, у новорожденных может отсутствовать. При резко выраженном шуме его диагностическое значение невелико, ибо он обычно лишен богатства оттенков и других характеристик, приобретаемых с возрастом. Одышка и цианоз у детей первых недель жизни наряду с пороками сердца могут служить проявлением респираторных, мозговых, метаболических и других нарушении.
Цианоз у новорожденного может быть вазомоторного происхождения, легочного, церебрального, метаболического и кардиального. Внимательный осмотр ребенка и простые тесты помогают разобраться в причине цианоза.
Вазомоторный цианоз наблюдается обычно на 2 — 3-й неделе жизни, реже — в течение первых месяцев. Для него характерна периферическая локализация (кисти, стопы). Губы и слизистые оболочки всегда остаются розовыми. При септикопиемии цианоз может приобретать генерализованный характер.
Легочный цианоз чаще связан с ателектазом, появляется вскоре после рождения, имеет генерализованный характер. Обычно этот вид синюхи уменьшается при крике ребенка и при ингаляции кислорода.
При гиалинозе легочных мембран заболевание начинается с одышки, а синюха достигает максимума через 24 — 48 ч. Как известно, чаще эту патологию можно наблюдать у недоношенных детей и у доношенных новорожденных, чьи матери больны сахарным диабетом.
Другие причины легочного цианоза — пневмония, диафрагмальная грыжа, трахеопишеводный свищ и агенезия легкого — распознаются с помощью общеклинических методов и рентгенологического исследования.
Церебральный цианоз наблюдается при отеке мозга, вызванном внутримозговым кровоизлиянием. Кроме специфического анамнеза, нередко отмечается характерный гемицианоз с четкой осевой демаркацией, что позволило иностранным авторам назвать этот симптом «костюм арлекина».
Метаболический цианоз встречается при тетании новорожденных, когда содержание кальция в сыворотке крови меньше 8 мг/100 мл и имеется гиперфосфатемия, а также при метгемоглобинемии. Дача глюконата кальция или метиленового синего помогает в распознавании этих форм синюхи.
Кардиальный цианоз имеет сбросовое происхождение, обычно носит тотальный характер и прогностически всегда серьезен. Кардиальный цианоз усиливается при крике, сопровождается одышкой, увеличением печени и ритмом галопа, сочетается с той или иной степенью недостаточности кровообращения. Последний синдром и артериальная гиноксемия являются основными причинами высокой летальности новорожденных с пороком сердца.
Анамнестическое и физикальное обследование новорожденного с подозрением на порок сердца проводится по общим правилам. Электрокардиография обязательна. При этом запись только стандартных отведений может дать ценную диагностическую информацию. Так, распознавание гипертрофии правого предсердия и правого желудочка возможно уже через 48 — 72 ч после рождения, а гипертрофии левых отделов — сразу после родов. Последний симптом наиболее часто указывает на врожденный порок сердца (трикуспидальная атрезия, атрезия легочной артерии с интактной межжелудочковой перегородкой). Для более детальной информации необходима запись грудных отведений с интерпретацией получаемых данных применительно к возрасту больного (М. Сандруччп, Г. Боно, 1966).
Рентгенологическое обследование позволяет оценить легочный рисунок, размер и конфигурацию тени сердца, характер его контуров. Трактовка рентгенограммы производится с учетом возрастных норм (В. И. Бураковский, Б. А. Константинов, 1970).
Наибольшую диагностическую ценность в плане распознавания пороков сердца и определения прогноза дают специальные инструментальные методы обследования — катетеризация полостей сердца и рентгеноконтрастная ангиокардиография. Показания к их применению ставятся при твердом убеждении в наличии у ребенка порока сердца.
Исследования выполняют под специальной премедикацией или в условиях наркоза. Катетер для записи давления, взятия проб крови и введения контрастного вещества (1,5 — 2 мл/кг) вводят через подкрыльцовую вену, большую подкожную вену бедра, основную бедренную вену пли через пупочные сосуды.
У новорожденных более безопасна и достаточна информативна селективная ангиокардиография, которая в подавляющем большинстве случаев позволяет обойтись без катетеризации полостей сердца. Только комплексное исследование больного ребенка с переходом по мере необходимости от более простых к более сложным методам дает возможность разобраться в сердечно-сосудистой патология.
Большинство врожденных пороков сердца имеет отчетливую возрастную хронологию первых клинических проявлений. В первые 4 недели жизни проявляются аномалии, при которых гемодинамика нарушается па ранних этапах плацентарного кровообращения (первая группа). К ним относят атрезию клапанных отверстий и недоразвитие того или иного отдела сердца.
Пороки второй группы наблюдаются у новорожденных, но могут проявиться и в более старшем возрасте. Эти пороки обусловливают острое нарушение кровообращения только после родов — в связи с началом легочного дыхания и увеличением мунутного объема (полная транспозиция сосудов, общий истинный артериальный ствол, двухкамерное сердце и др.).
Пороки третьей группы у новорожденных не выявляются. Сюда относят дефекты межпредсердной, межжелудочковой п аорто-легочной перегородок и др. При септальных дефектах развертыванию клинической картины у новорожденных препятствует высокое легочное артериолярное сопротивление. Так, при дефектах межжелудочковой перегородки сброс крови возникает по мере снижения резистентности сосудов легких, т. е. начиная с двухмесячного возраста, когда и появляются первые симптомы заболевания. При тетраде Фалло в течение 1-го месяца действуют механизмы, связанные с возрастными особенностями и препятствующие
развитию патологического эффекта (постэмбриональная полиглобулия, низкий основной обмен и т. п.).
К четвертой группе относят пороки, при которых нарушения в гемодинамике могут проявляться в любом возрасте в зависимости от анатомического варианта.
Выделение четырех категорий суживает диапазон поисков при топической диагностике пороков сердца у новорожденных (табл. 16).
Таблица 16. Разделение врожденных пороков сердца в зависимости от возрастной хронологии первых симптомов
Пороки, проявляющиеся только у новорожденных |
Пороки, проявляющиеся преимущественно у новорожденных |
Пороки, не проявляющиеся у новорожденных |
Пороки, встречающиеся во всех возрастных группах |
1. Гипоплазия левой половины сердца |
1. Полная транспозиция аорты и легочной артерии |
1. Дефект межпредсердной перегородки |
1. Открытый артериальный проток |
2. Атрезия клапанов легочной артерии с интактной межжелудочковой перегородкой |
2. Общий истинный артериальный ствол |
2. Дефект межжелудочковой перегородки |
2. Коарктация аорты |
3. Поддиафрагмальный полный аномальный дренаж легочных вен |
3. Двухкамерное сердце |
3. Тетрада Фалло (кроме случаев с атрезней легочной артерии) 4.4. Аномальное отхождение левой венечной артерии от легочной |
3. Стеноз клапанов легочной артерии 4. Стеноз аорты 5. Тетрада Фалло с атрезией легочной артерии 6. Атрезия трикуспидального клапана 7. Единственный желудочек 8. Болезнь Эбштейна 9. Полный (наддиафрагмальный) аномальный дренаж легочных вен 10. Атрио-вентрикулярный канал |
Удельный вес и значение отдельных врожденных пороков сердца неодинаковы в различные возрастные периоды. Поэтому в данной главе мы приводим описания клинической картины пороков сердца, наиболее часто встречающихся у новорожденных.
Полная транспозиция аорты и легочной артерии
Полная транспозиция аорты и легочной артерии — один из наиболее распространенных пороков сердца у новорожденных. По секционным данным, его частота варьирует от 8,4 до 37,5% от числа всех случаев заболеваний сердечно-сосудистой системы у новорожденных (Б. А. Константинов, 1969; Donzelot, Dallaines, 1954). Порок этот определяют, как «самую частую причину цианоза и сердечной недостаточности» младенцев или как «самый частый порок, требующий операции» у ребенка первых недель и месяцев жизни (Cooley, 1966).
При полной транспозиции сосудов аорта отходит от правого желудочка, располагаясь впереди устья легочной артерии. Последняя берет начало от левого желудочка и прилежит к аорте сзади.
Газообмен и, следовательно, жизнь больного возможны лишь при наличии дополнительных коммуникаций между большим и малым кругами кровообращения. Чаще всего это открытое овальное окно, дефект межжелудочковой перегородки, дефект межпредсердной перегородки или открытый артериальный проток. Через эти сообщения происходит обменный сброс крови, и только таким образом часть окисленной крови может попадать в аорту, а часть венозной — в легкие. Величина сброса при транспозиции сосудов определяется размерами сообщений, величиной сосудистой резистентности и другими факторами. Количество окисленной крови, поступающей в аорту у таких больных, невелико, поэтому сразу после рождения ребенок начинает испытывать тяжелейшую артериальную гипоксемию, равной которой по степени не бывает при других пороках. Мы наблюдали новорожденных, у которых насыщение артериальной крови кислородом составляло 20 — 40% НbO2.
Вскоре после рождения у больных значительно возрастает объем циркулирующей крови как компенсация тканевой гипоксии. Это неизбежно ведет к перегрузке сердца и развитию недостаточности кровообращения.
Таким образом, для гемодинамики при полной транспозиции сосудов характерно разобщение кругов кровообращения, при котором правый желудочек выполняет роль насоса для большого круга. Газообмен осуществляется при помогли переменных сбросов крови через дополнительные коммуникации.
Клиническая картина. Большой минутный объем сердца ведет к его перегрузке и развитию недостаточности кровообращения. Порок встречается преимущественно у мальчиков. Дети рождаются с нормальным весом. Первый симптом — синюха — появляется с первого дня рождения, носит интенсивный, стойкий и общий характер. В 1 — 2-ю нед одышки не наблюдается, сердце не увеличено, конфигурация его обычная. Отеков, увеличения печени и других признаков недостаточности кровообращения в это время не отмечается. С 3 — 4-й нед жизни появляются тахипноэ, затруднения при кормлении, тахикардия с ритмом галопа, увеличивается печень, появляются хрипы в легких, периферические отеки, асцит. Быстро увеличиваются размеры сердечной тени, которая по конфигурации все больше напоминает косо расположенный овал или «яйцо, лежащее на боку» («egg on theside» английских авторов).
Кажущееся «благополучие» у синюшного новорожденного в первые 2 — 3 нед после рождения, сменяющееся быстро наступающей и неудержимо прогрессирующей сердечной декомпенсацией — характерный признак полной транспозиции сосудов.
Однако ждать развития декомпенсации у новорожденного с цианозом и на этой основе ставить диагноз — нельзя. Если учесть, что средняя продолжительность жизни у ребенка с полной транспозицией сосудов составляет 3 мес (Hanlon, Blalock, 1948), то становится понятным, как мало у врача времени для постановки топического диагноза и как он важен в первые 2 — 3 нед после рождения. Объективное обследование, кроме резкого и тотального цианоза, позволяет выявить сердечный горб, у ряда больных — систолический шум слева от грудины. Более характерна полная или почти полная афоничность. Симптомы недостаточности кровообращения, как было сказано выше, присоединяются с 3 — 4-й нед жизни.
Электрокардиографическое исследование демонстрирует наличие право- граммы с выраженной гипертрофией правых отделов сердца. Перегрузка правого желудочка обычно не определяется.
Рентгенологическая картина свидетельствует о нормо- или гиперволемии легких и нормальных размерах сердца. С 3 — 4-й нед жизни тень сердца увеличивается влево, подчеркивая узость сосудистого пучка в фасной проекции, и все больше начинает напоминать овал (рис. 52).
Рис. 52. Рентгенограмма грудной клетки ребенка с полной транспозицией аорты и легочной артерии, открытым овальным окном. Силуэт сердца увеличен и напоминает по форме овал.
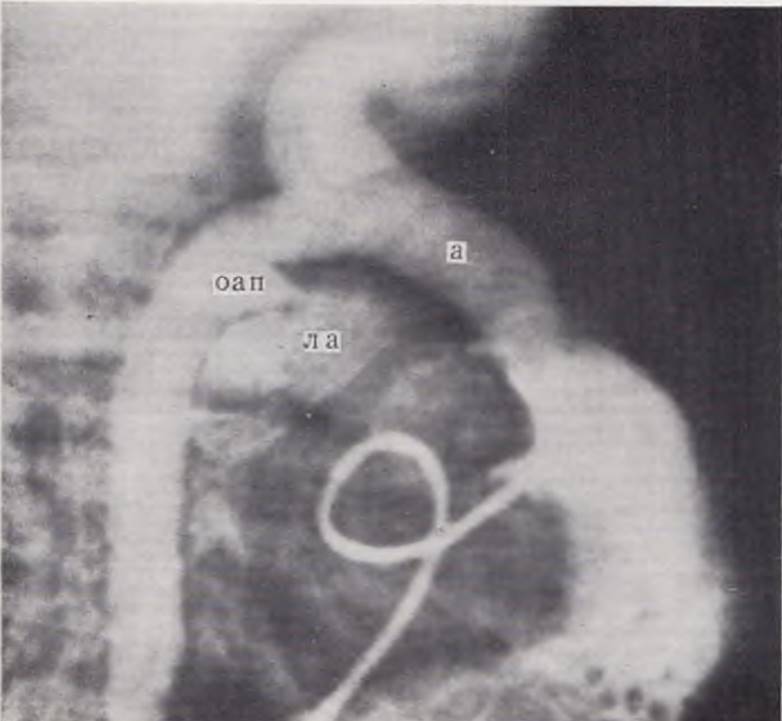
При подозрении на данный порок ребенку показана селективная рентгеноконтрастная ангиокардиография из полости правого желудочка с обязательной съемкой в двух проекциях. Достоверным признаком порока является изображение аорты, берущей начало от правого желудочка и располагающейся впереди легочной артерии. При этом бульбус аорты расположен необычно высоко и как бы кпереди от сердечной тени (рис. 53).
Рис. 53. Селективная ангиокардиограмма ребенка с полной транспозицией аорты и легочной артерии, открытым овальным окном, незаращением артериального протока. Из правого желудочка контрастируется аорта (а), лежащая кпереди от легочной артерии (ла), последняя заполняется через боталлов проток (оап).
Дифференциальная диагностика порока отражена в табл. 17.
Таблица 17. Дифференциальная диагностика важнейших поражений сердца у новорожденных
Диагноз |
Время первых проявлений заболевания от рождения |
Первый симптом |
Одышка |
Цианоз |
Пульс |
ЭКГ |
Увеличение сердца |
Недостаточность кровообращения |
Усиление легочного рисунка |
Примечание |
|
1. Гипоплазия левой половины сердца |
Первые сутки |
Внезапная одышка |
+++ |
+ |
Не пальпируется |
Правограмма |
+ + + |
+++ |
+++ |
Встречается у мальчиков в 2 раза чаще, 75% погибает в 1-ю неделю жизни |
|
2. Атрезия клапанов легочной артерии |
С рождения |
Цианоз |
+++ |
++ |
Обычный |
Перегрузка левых отделов |
++ |
+++ |
— |
||
3. Поддиафрагмальный дренаж легочных вен |
24 ч — 7 дней |
» |
++ |
++ |
Слабый |
Правограмма |
— |
++ |
+++ |
||
4. Транспозиция сосудов |
С рождения |
Цианоз |
+ |
+ + + |
Обычный |
Правограмма |
++ |
++ |
— или ++ |
Силуэт сердца в виде «яйца» на боку. У мальчиков встречается в 2 1/2раза чаще |
|
5. Коарктация аорты |
Любое |
Одышка |
++ |
— |
Усилен на руках |
Правограмма |
++ |
++ |
— или + |
||
6. Атрезия трехстворчатого клапана |
С рождения |
Цианоз |
+ — |
++ |
Обычный |
Левограмма |
+ |
— |
— или + |
||
7. Общий артериальный ствол |
1 — 4 нед |
Одышка |
++ |
+ — |
Скорый, полный |
Комбинированная гипертрофия |
+ |
+ — |
+ + или — |
||
8. Тетрада Фалло с атрезией легочной артерии |
1 — 4 нед |
Цианоз |
+ — |
Обычный |
Правограмма |
+ |
— |
— |
|||
Комбинированная гипертрофия |
|||||||||||
9. Боталлов проток |
Любое |
Одышка |
++ |
— |
Скорый, полный |
+ |
++ |
++ |
|||
10. Болезнь Эбштейна |
» |
Цианоз |
+ |
++ |
Обычный |
Характерны низковольтные расщепленные комплексы |
++ |
++ |
— |
||
11. Миокардит |
» |
Тахикардия |
++ |
+ — |
Слабый |
++ |
++ |
— |
|||
12. Гиалиноз легочных мембран |
Первые 6 ч после рождения |
Одышка |
+++ |
+ |
» |
+ |
++ |
++ |
Особенно часто у недоношенных |
Условные обозначения: — отсутствие симптома, + — непостоянный симптом, + умеренно выраженный симптом, + + значительно выраженный симптом, +++ резко выраженный симптом.
Лечение. Новорожденный с транспозицией сосудов нуждается в незамедлительной терапии. Лечение включает кислородотерапию и назначение сердечных гликозидов. К оперативному лечению следует прибегать как можно быстрее после установления диагноза.
Применяют различные паллиативные вмешательства, направленные на увеличение переменных сбросов крови с целью уменьшения артериальной гипоксемии.
В последнее время большое распространение находит закрытая атриосептостомия, выполняемая непосредственно в кабинете зондирования. Показанием к этой операции, которая производится без торакотомии, служит насыщение артериальной крови кислородом ниже 75% НbО2(Rashkind, Miller, 1966).

С целью увеличения размеров межпредсердного сообщения больному через одну из периферических вен в сердце проводят катетер типа Фоггарти. После того как оперирующий убедился в том, что катетер находится в левом предсердии, с помощью рентгеноконтрастного вещества раздувают расположенный на конце зонда баллон. Далее потягиванием за катетер межпредсердную перегородку подводят к устью нижней полой вены и резким движением извлекают катетер из левого предсердия. При этом межпредсердная перегородка разрывается и через образованное отверстие устанавливается сброс крови (рис. 54).
Рис. 54. Схема закрытой атриосептостомии. Проведение катетера в левое предсердие и раздувание баллончика (а, б). Низведение межпредсердной перегородки к нижней полой вене (в), резкое извлечение катетера, образование дефекта в перегородке сердца (г).
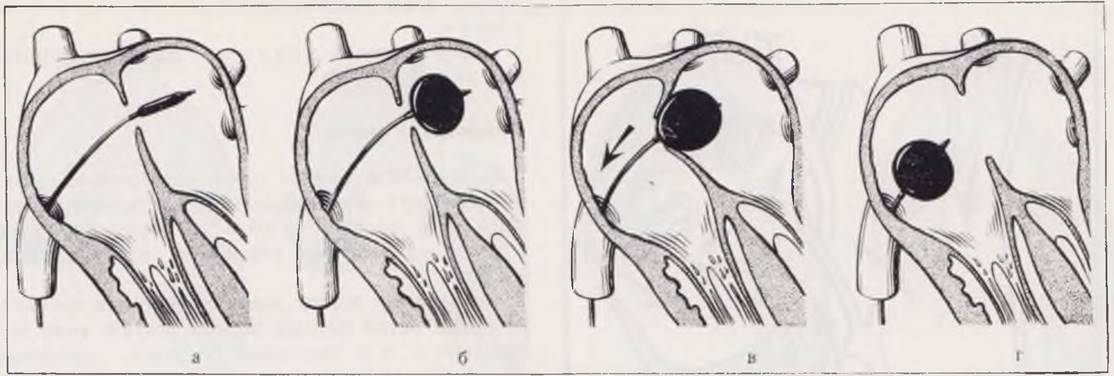
Обычно эффект наступает немедленно и проявляется уменьшением или даже исчезновением цианоза, повышением периферического артериального насыщения крови кислородом (Б. А. Константинов, 1969; Ю. С. Петросян, В. А. Гарибян, 1972). Другие операции — атрио- септэктомия, межартериальный анастомоз, суживание легочной артерии — у новорожденных выполняются редко.
Гипоплазия левой половины сердца
Гипоплазия левого сердца является одним из наиболее распространенных пороков, составляя 7 — 15% всех врожденных аномалий сердца (В. П. Жуковский, 1913; Noon, Nadas, 1958). Наблюдается она исключительно у новорожденных, так как 75% больных погибают в 1-ю неделю жизни, а остальные — в течение 1-го месяца. Порок представляет собой сложный клинико- морфологический комплекс — атрезию восходящей аорты и гипоплазию левого желудочка. Это собирательное понятие включает также предродовое заращение овального окна, атрезию или резкий стеноз митрального клапана, которые обычно встречаются в ассоциации друг с другом и ведут к недоразвитию левых отделов сердца и аортального клапана.
В происхождении порока играет роль уменьшение притока крови через овальное окно к левому желудочку и повышенный ее сброс через боталлов проток во время плацентарного кровообращения. Общими морфологическими признаками синдрома недоразвития левого сердца являются: 1) резкая гипоплазия левых отделов
сердца, его клапанного аппарата и аортального тракта; 2) гипертрофия правых отделов с большой легочной артерией и зияющим боталловым протоком; 3) вторичный эндокардиальный фиброэластоз.
Восходящая аорта резко сужена, просвет ее сохранен, клапаны полностью сращены между собой. Митральное кольцо имеет небольшой диаметр, створки его недоразвиты или сращены. Левый желудочек имеет толстые стенки и полость в виде узкой щели. Эндокард плотный, грубый, белесовато-желтого цвета.
Гемодинамика нарушается на ранних этапах плацентарного кровообращения. Путь кровотока при этом можно представить следующим образом: полые вены —» правое предсердие —» правый желудочек — легочная артерия —» боталлов проток —» дуга аорты (ретроградно) —» нисходящая аорта (рис. 55).
Рис. 55. Гемодинамика при гипоплазии левого сердца.
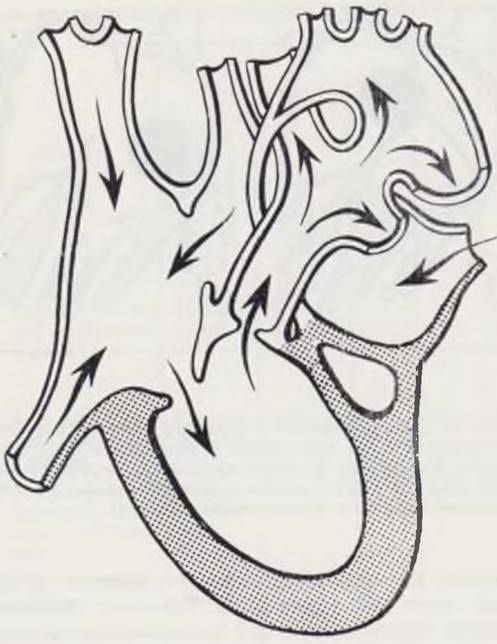
После рождения направление циркуляции не меняется. Кровь в легкие нагнетается под большим давлением, в то время как ее отток по венам затруднен из-за малой емкости левых отделов. Поэтому с рождения устанавливается артериальная легочная гипертония с венозным застоем. Из левого предсердия кровь может оттекать только в правое, где перемешиваются артериальный и венозный потоки. Обязательным условием для жизни ребенка является наличие у него в дополнение к боталлову протоку открытого овального окна или аномального дренажа легочных вен. Всю нагнетательную функцию выполняет правый желудочек. Гемодинамика при гипоплазии левой половины сердца характеризуется наличием одного функционирующего (правого) желудочка, легочной гипертензией с венозным застоем, артериальной гипоксемией.
Клиническая картина. Порок наблюдается чаще у мальчиков. Заболевание проявляется внезапным началом — одышкой в первые сутки после рождения. Тахипноэ сопровождается прогрессирующей недостаточностью кровообращения. Синюшность выявляется позже и, хотя носит общий характер, никогда не достигает значительных степеней (Noon, Nadas, 1958).
Объективное обследование выявляет резко учащенное (80 — 100 в минуту) и аритмичное дыхание с западанием уступчивых мест грудной клетки и раздуванием крыльев носа. Обращает на себя внимание слабая пульсация периферических артерий, не улавливаемая даже на сонных сосудах. В большинстве случаев порок афоничен.
Со стороны сердца отмечается эмбриокардия, в легких с первых дней определяется бронхиальное дыхание, обилие разнокалиберных хрипов. Печень быстро увеличивается, отеки и асцит не успевают развиться.
На электрокардиограмме преобладает правограмма. Реже наблюдается левограмма с комбинированной гипертрофией. В этих случаях на вскрытии находят фиброэластоз левого желудочка.
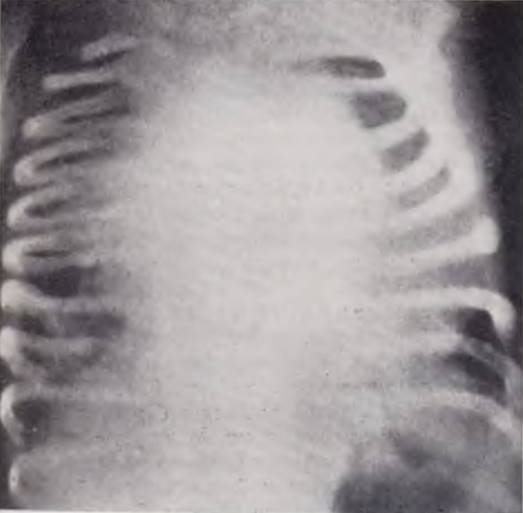
При рентгенологическом исследовании сердце увеличено уже в первые часы после рождения. Тень его, неправильно округлая, прогрессивно растет (рис. 56). Рентгеноконтрастное изучение выявляет большие размеры правого желудочка и нисходящую аорту, являющуюся прямым продолжением боталлова протока. После легочной капиллярной фазы контрастируются правые отделы. Изображения левых камер и восходящей аорты получить не удается.
Рис. 56. Рентгенограмма грудной клетки ребенка с гипоплазией левого сердца и атрезией восходящей аорты. Силуэт сердца неправильной формы, резко увеличен в размерах.
Эффективного лечения до сегодняшнего дня не найдено.
Коарктация аорты
Порок представляет собой врожденное сужение просвета аорты вплоть до его полного перерыва, как правило, в области перешейка дуги у места отхождения открытого артериального протока.
Коарктация аорты довольно часто встречается среди детей первых недель жизни, хотя наблюдается и у взрослых больных, составляя около 10 — 15% всех врожденных заболеваний сердечно-сосудистой системы (Mehrizi, 1964). По данным Sloan и Cooley (1953), коарктация аорты встречается один раз на 500 вскрытий. У мальчиков она наблюдается вдвое чаще (Campbell, Polani, 1961).
Анатомия порока может значительно варьировать, чем и объясняется большое количество предложенных классификаций.
Для практического врача достаточно различать два основных типа порока — постдуктальный и преддуктальный (рис. 57).
Рис. 57. Классификация различных вариантов коарктации аорты: постдуктальная (взрослая) коарктация (а). Преддуктальная (инфантильная) коарктация (б, в, г).
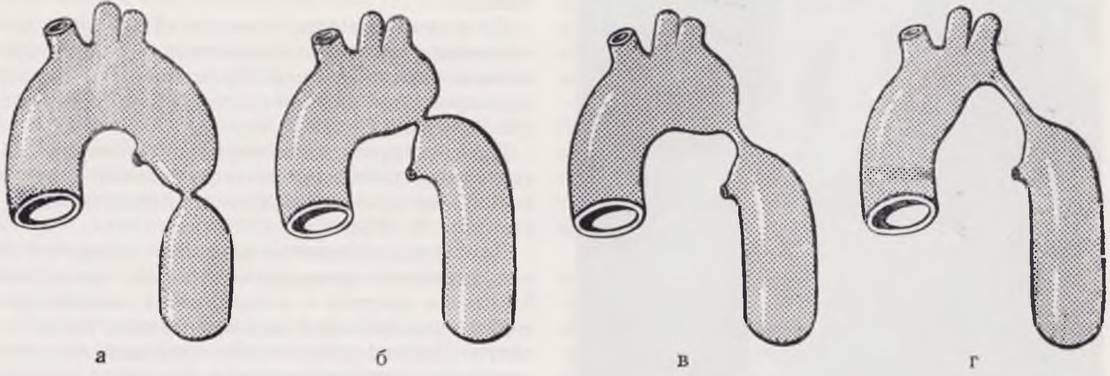
При постдуктальной, или «взрослой», коарктации аорты сужение располагается дистальнее устья боталлова протока или артериальной связки и обычно локализовано по протяжению. У больных всегда наблюдается гипертрофия левого желудочка, а около 25% пациентов имеют двустворчатый клапан аорты. Нарушение кровообращения обусловлено наличием механического препятствия. В обход места сужения рано развиваются коллатеральные сосуды. В верхней и нижней половинах организма возникают два различных режима кровообращения. Артериальный проток может быть заращен или функционирует.
При преддуктальной, или «инфантильной», коарктации аорты сужение располагается проксимальнее устья боталлова протока. Последний обычно имеет большой диаметр и является как бы продолжением легочной артерии в нисходящую аорту. Преддуктальная коарктация аорты может быть локальной, протяженной и распространяться на дугу аорты.
При последнем варианте в 80% наблюдений имеются дополнительные тяжелые пороки сердца — дефект межжелудочковой перегородки, атриовентрикулярный канал, единственный желудочек, транспозиция сосудов и др. (Keith е. а., 1968). При инфантильном типе коарктации аорты гипертония в большом круге кровообращения не развивается, коллатеральное кровообращение отсутствует. В то же время у пациентов с этим вариантом порока устанавливается сброс крови справа налево: из легочной артерии в нисходящую аорту. При этом правое сердце испытывает резкую перегрузку, так как принимает весь минутный объем левого сердца и дополнительно часть своего выброса, равную величине шунта. Малый круг кровообращения характеризуется высоким артериальным давлением. Таким образом, при «взрослом» типе порока имеет место артериальная гипертония верхней половины тела, а при «инфантильном» — легочная гипертония.
Клиническая картина. Коарктация аорты проявляется клинически на 1-м году жизни примерно у половины детей. Около 2/3 из них имеют преддуктальный тип коарктации.
Несмотря на принципиальное различие в гемодинамике и в прогнозе у больных с преддуктальным и постдуктальным вариантами коарктации аорты, симптоматика у них весьма сходна. В подавляющем большинстве случаев коарктация аорты у младенцев напоминает длительное респираторное заболевание с прогрессирующей недостаточностью кровообращения.
Примерно в 80% всех наблюдений первым симптомом коарктации аорты является одышка (Marks е. а., 1953), которая может возникнуть уже спустя 48 — 72 ч после рождения ребенка. В других случаях порок проявляется затруднением при кормлении, раздражительностью, кашлем, отставанием в весе. В ряде случаев родители отмечают у ребенка болевые приступы (типа абдоминальных колик). Объективное обследование позволяет отметить обычную окраску кожных покровов и видимых слизистых оболочек.
При преддуктальной коарктации аорты в ряде случаев удается выявить цианоз нижней половины тела. Одышка всегда резко выражена, межреберья западают. При постдуктальном варианте уже в первые недели жизни можно отметить превалирующее развитие верхнего плечевого пояса.
Примерно у половины всех больных шума прослушать не удается, у другой половины он имеет систолический характер, разнообразную интенсивность и локализацию. Часто шум бывает обусловлен дефектом межжелудочковой перегородки или другим сопутствующим пороком.
В большинстве случаев к одышке присоединяются другие симптомы сердечной недостаточности: увеличение границ сердца, увеличение печени, периферические отеки. Хрипы в легких застойного характера определяются не часто и обычно в терминальной фазе заболевания.
Важную информацию дает пальпация пульса и измерение давления на руках и ногах. Пульс на руках обычно нормальный или усиленный, а на ногах отсутствует. При преддуктальной коарктации пульс на ногах сохраняется. Артериальное давление на верхних конечностях может достигать 180 — 200 мм рт. ст. При постдуктальном варианте давление на ногах равно нулю.
При электрокардиографическом обследовании детей первых 2 мес жизни чаще наблюдается правограмма при обоих анатомических вариантах. Важное диагностическое значение электрокардиограмма имеет в случаях, когда уже в первые педели жизни определяется левограмма или нормальная электрическая ось, что дает основание предположить у ребенка постдуктальную коарктацию аорты.
При рентгенологическом исследован и и находят увеличение сердечной тени и усиление легочного рисунка. Узурация ребер и другие рентгенологические признаки, характерные для коарктации у взрослых, появляются после первого года жизни. Конфигурация всей сердечной тени и ее контуров может значительно варьировать.
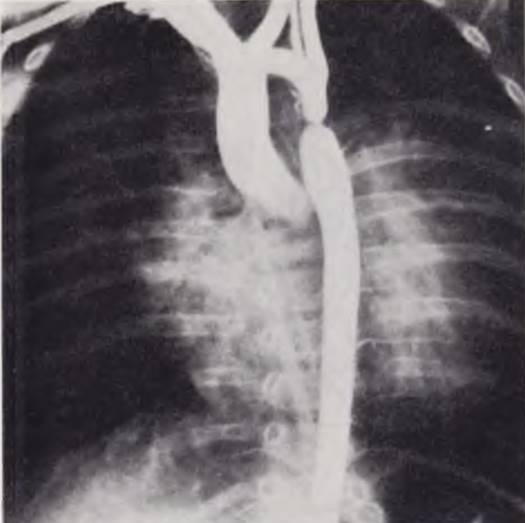
Коарктация аорты должна быть заподозрена у младенца с недостаточностью кровообращения, резистентной к сердечным гликозидам, и с разницей пульса и артериального давления на руках и йогах. В этих случаях можно обойтись без катетеризации полостей сердца и аортографии. В неясных случаях показано зондирование сердца и рентгеноконтрастное исследование для уточнения диагноза и выявления сопутствующих пороков (рис. 58).
Рис. 58. Аортограмма ребенка с коарктацией аорты постдуктального типа, недостаточность кровообращения НБ степени, недостаточность внешнего дыхания.
Прогноз при данном пороке у младенцев всегда серьезен. Если не проводится лечение, на первом году жизни погибает 80% детей с недостаточностью кровообращения (Gammelguard е. а., 1959). Особенно серьезен прогноз при преддуктальном варианте коарктации из-за большой распространенности сопутствующих
пороков и необратимости легочной гипертонии.
Лечение во всех случаях начинают с назначения сердечных гликозидов. Однако при данном пороке они малоэффективны и только операция дает возможность ребенку выжить (В. И. Францев, 1966).
При постдуктальном варианте резекция коарктации с последующим анастомозом конец в конец дает стойкое излечение. Летальность составляет 2 — 8%.
При преддуктальном варианте предложена паллиативная операция — сужение зияющего боталлова протока с последующей окончательной его перевязкой и восстановлением просвета аорты. Однако после таких операций до сегодняшнего дня наблюдается высокая летальность.
Открытый артериальный проток
Открытый артериальный проток принято считать врожденным, если он продолжает функционировать у ребенка спустя неделю после рождения (Gasul, 1966). Проток диаметром более 2 см не способен к спонтанному закрытию. Данная аномалия хорошо известна практическим врачам, так как является весьма распространенной и составляет от 10 до 30% всех врожденных пороков сердца (Abbott, 1936). У детей старшего возраста гемодинамика и клиническая картина открытого артериального протока изучены подробно (Б. В. Петровский, А. А. Кешишева, 1963; Ф. X. Кутушев, Л. Р. Плотникова, 1967).
Течение данного порока у новорожденных отличается большими особенностями, главными из которых является наличие недостаточности кровообращения (Taussig, 1955). Открытый артериальный проток дает ранние тяжелые клинические проявления у каждого восьмого больного (Рrее е. а., 1962).
Описываемый порок нередко встречается в комбинации с другими, однако в данной главе речь пойдет только о случаях изолированного боталлова протока.
Гемодинамика при открытом артериальном протоке в пренатальном периоде подробно освещена (В. И. Бураковский, Б. А. Константинов, 1970) и здесь рассматриваться не будет. В патологических ситуациях через него устанавливается сброс крови из аорты в малый круг кровообращения. Величина этого шунта определяется диаметром протока и соотношением сосудистого сопротивления в большом круге кровообращения и в легких.
В первые недели жизни сброс происходит преимущественно во время систолы, так как диастолический градиент между кругами кровообращения в этот период невелик. По мере снижения легочной сосудистой резистентности шум возрастает и начинает занимать весь сердечный цикл (Rudolph е. а., 1958). Артериовенозный сброс крови переполняет малый круг, перегружает левые отделы сердца, снижает эффективность легочного кровотока, в связи с чем возникает декомпенсация кровообращения.
Гемодинамика при открытом артериальном протоке характеризуется легочной гиперволемией, перегрузкой левых отделов сердца и развитием недостаточности кровообращения по левому типу.
Клиническая картина. Симптомы порока у новорожденного напоминают подострое респираторное заболевание, к которому присоединяется недостаточность кровообращения. Первые признаки — затруднения при кормлении, субфебрильная температура, кашель могут проявиться с 3 — 4-го дня жизни. В первые дни обычно ставится диагноз пневмонии, бронхита. Объективное исследование выявляет прогрессирующую недостаточность кровообращения — одышку, тахикардию, увеличение печени, хрипы в легких. Ребенок при крике может синеть (особенно нижняя половина тела), так как в этом возрасте легко возникает временный обратный сброс крови.
При аускультации шум либо не определяется, либо выслушивается короткий систолический шум слева от грудины. Сердце всегда увеличено, легочный рисунок усилен. При электрокардиографическом исследовании чаще определяется нормальная электрическая ось и комбинированная гипертрофия желудочков.
Основные симптомы незаращения артериального порока выявляются при изучении пульса п пульсового давления (Krovetz, 1962). При этом характерны скорый и высокий пульс (pulsus celer et altus) и пульсовое давление, превышающее 40 мм рт. ст.
Прогноз во многом определяется возрастом, в котором впервые появляется недостаточность кровообращения. У новорожденных функциональное закрытие протока в огромном большинстве случаев происходит в течение 1-х суток, тогда как анатомическая облитерация заканчивается ко 2 — 3-му мес жизни (Gessner, 1965).
Лечение. Терапию необходимо начинать незамедлительно с дигитализации. В ряде случаев после этого с целью уточнения диагноза можно выполнить аортографию путем пункции подкрыльцовой артерии (Г. И. Алексеев, 1968).
Консервативная терапия при недостаточности кровообращения у новорожденных с открытым артериальным протоком часто неэффективна. Pate и Ainger (1963) сообщили о смерти 44 грудных детей из 45, леченных дигиталисом. В таких случаях мы рекомендуем раннюю операцию. Методом выбора является перевязка протока двумя шелковыми лигатурами. Операция приводит к выздоровлению.
Атрезия клапанов легочной артерии с интактной межжелудочковой перегородкой
При этом пороке правый желудочек не имеет выхода и отделен от легочного русла клапанами, сросшимися в сплошную мембрану, или мышечной перемычкой. Аномалия встречается примерно в 0,8% всех врожденных пороков сердца, причем почти исключительно у детей первых недель жизни (Gasnl, 1966).
Различают два варианта порока (рис. 59). При первом, который встречается в подавляющем большинстве наблюдений, правый желудочек имеет минимальную внутреннюю полость и резко утолщенные стенки. При втором варианте полость правого желудочка резко увеличена, трикуспидальное отверстие зияет.
Рис. 59. Схема кровообращения при атрезии легочной артерии с интактной межжелудочковой перегородкой при малой полости правого желудочка (а), при большой полости правого желудочка (б).
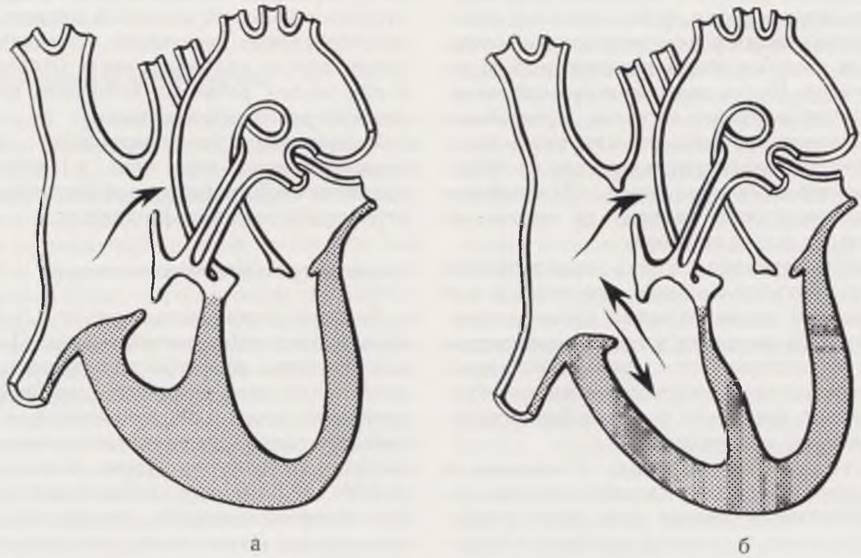
Гемодинамика нарушена уже с ранних этапов плацентарного кровообращения. При небольшой полости правого желудочка кровь из правого предсердия поступает через овальное окно в левое сердце, артериальный желудочек резко гипертрофируется. При выраженной полости венозного желудочка всегда имеется трикуспидальная регургитация, за счет которой этот желудочек периодически опорожняется. Далее, как и при первом варианте, кровь сбрасывается в левое сердце через овальное окно. Легочная циркуляция у новорожденного осуществляется за счет боталлова протока и бронхиальных сосудов. После рождения правый желудочек с блокированным выходом продолжает обычно поддерживать давление в венозном предсердии выше, чем в артериальном, что не позволяет овальному окну закрыться. Дальнейшая судьба новорожденного зависит от сроков закрытия артериального протока.
При первом варианте порока гемодинамика напоминает трикуспидальную атрезию, а при втором — резкий изолированный стеноз легочной артерии. Оба варианта в гемодинамическом отношении характеризуются хронической артериальной гипоксемией, прогрессирующим дефицитом легочного кровотока и одним функционирующим (левым) желудочком.
Клиническая картина. Основным и первым симптомом порока является цианоз, который появляется в первые дни после рождения ребенка, носит тотальный характер и быстро прогрессирует. К синюхе довольно быстро присоединяется одышка. Спустя неделю появляются признаки недостаточности кровообращения по правому типу — гепатомегалия, периферические отеки, ритм галопа по верхушке сердца. Особенно характерно раннее наступление декомпенсации при варианте с большой полостью правого желудочка и трикуспидальной недостаточностью. В этих случаях нередко можно отметить пульсацию яремных вен и печени.
Для описываемого порока характерно умеренное увеличение кардиальных размеров в первые дни и прогрессивное увеличение в последующие. Порок чаще всего афопичен.
На электрокардиограмме электрическая ось может быть отклонена вправо, влево и быть нормальной. Постоянно наблюдается увеличение зубца Р2. В грудных отведениях при первом варианте чаще встречаются гипертрофия и перегрузка левых отделов сердца, а при втором — возможна комбинированная гипертрофия.
Рентгенологическое исследование выявляет ту или иную степень увеличения размеров сердца, силуэт которого очень вариабелен, и обеднение легочного рисунка.
Основное место в топическом распознавании порока принадлежит селективной ангиокардиографии из полости правого предсердия.
Дифференциальный диагноз труден (см. табл. 17). Прогноз при легочной атрезии серьезен. Около одной трети детей умирают к концу 2-й нед жизни, а половина — к концу 1-го месяца (Keith е. а., 1968).
Лечение хирургическое. Из отдельных видов оперативного вмешательства при легочной атрезии с большой полостью правого желудочка целесообразнее выполнять закрытую трансвентрикулярную вальвулотомию (Robinson, 1965), а при малых размерах венозного желудочка — межартериальный анастомоз.
Возможно, у новорожденных с небольшими размерами овального окна в качестве первой паллиативной процедуры эффективной окажется атриосептостомия по Рашкинду.
Атрезия трехстворчатого клапана
Данный порок составляет от 1,5 до 3% всех врожденных заболеваний сердца. Правое венозное отверстие при этом пороке полностью заращено и венозное предсердие сообщается с артериальным через открытое овальное окно. Правый желудочек резко гипоплазирован и может сообщаться с левым через межжелудочковый дефект. В 80% всех наблюдений имеется стеноз легочной артерии, атрезия ее ствола или гипоплазия. Очень часто прп трикуспидальной атрезии встречается транспозиция аорты и легочной артерии. Легочный кровоток осуществляется за счет боталлова протока.
Гемодинамика очень сходна с кровообращением при атрезии легочной артерии.
Клиническая картина. У большинства больных цианоз появляется уже в первую неделю жизни. Синюшность обычно резко выражена. Умеренная и поздняя синюха характерна для больных, которые имеют большое межпредсердное сообщение и стеноз легочной артерии. Помимо цианоза, обращает на себя внимание левограмма, гипертрофия левых отделов на электрокардиограмме с блокадой левой ножки пучка Гиса. Сердце не увеличено, явления декомпенсации сравнительно долго отсутствуют. Несмотря па последнее обстоятельство, нетрудно уловить пульсацию шейных вен и пульсацию края печени. Аускультация не характерна.
Диагностика у новорожденного с выраженным цианозом с перегрузкой левых отделов на электрокардиограмме при небольших размерах сердца и с характерным свободным ретростернальным пространством на рентгенограмме, снятой во второй косой позиции, практически не сложна (см. табл. 18). Во многих случаях порока ангиокардиография для подтверждения диагноза не требуется. Прогноз тем серьезнее, чем раньше и интенсивнее выражен цианоз.
Лечение. У новорожденных в последнее время оказалась эффективной паллиативная операция — атриосептостомия по Рашкинду (Ю. С. Петросяп, В. А. Гарибян, 1972), увеличивающая диаметр межпредсердного сообщения и значительно уменьшающая степень артериальной гипоксемии.
Аномальный дренаж легочных вен
Аномальный дренаж легочных вен в венозную систему большого круга кровообращения встречается у 1,5 — 3% всех новорожденных с врожденными заболеваниями сердца и сосудов (Ф. II. Ромашов, 1965; Smith е. а., 1961).
Нередко аномальный дренаж легочных вен наблюдается при таких сложных пороках, как двухкамерное сердце, единственный желудочек, транспозиция сосудов и общий артериальный ствол. В этих случаях у новорожденных имеется синдром асплении. Эти комбинации несовместимы с длительным выживанием.
Для практического врача больший интерес представляют так называемые изолированные формы полного аномального дренажа легочных вен. Их анатомия весьма разнообразна. Наиболее исчерпывающую классификацию форм порока в зависимости от уровня впадения аномальных вен в систему большого круга кровообращения представили Dakling с соавт. (1957). Авторы различают в порядке убывающей частоты четыре анатомические формы порока: 1) супракардиальную (встречается в половине всех случаев полного аномального дренажа легочных вен); 2) кардиальную; 3) инфракардиальную; 4) смешанную.
При супракардиальной форме устья легочных вен впадают в левую или правую верхнюю полую вену. При кардиальной форме они дренируются в коронарный синус или правое предсердие, при инфракардиальной — в нижнюю полую вену, в портальную вену или венозный проток. Наконец, при смешанной форме устья легочных вен могут открываться одновременно в коропарный синус и в левую безымянную вену.
Несмотря на разнообразие анатомических вариантов, гемодинамика при описываемом пороке имеет много общего. Она характеризуется резкой легочной плеторой при низкой эффективности кровотока в малом круге кровообращения. Минутный объем большого круга снижен, причем его величина зависит от объема сброса крови справа налево на уровне межпредсердного сообщения (чаще всего через открытое овальное окно). Для порока характерны также умеренная артериальная гипоксемия, резкая перегрузка правых отделов сердца, раннее развитие недостаточности внешнего дыхания и кровообращения. С первых дней жизни ребенка существует легочная гипертония.
Клиническая картина. Первые проявления порока в виде одышки, кашля и затруднений при кормлении встречаются у подавляющего большинства детей в первые недели и месяцы жизни. При инфракардиальном варианте порока клинические признаки появляются с первых дней жизни. Рано выявляются цианоз и недостаточность кровообращения. Правда, синюха никогда не достигает резких степеней и всегда уступает главенствующее место одышке. При инфракардиальных формах наблюдается очень характерный диагностический симптом: несмотря на резкую декомпенсацию, размеры сердца остаются нормальными. При других вариантах порока, как было сказано выше, начало не такое острое. Многие дети с момента рождения не покидают больничных учреждений из-за бесконечной вереницы «респираторных» заболеваний.
Объективное исследование выявляет резкую гипотрофию, легкий, временами усиливающийся цианоз, выраженный сердечный горб. Сердце увеличено в размерах, во втором межреберье слева выслушиваются систолический шум, акцепт II топа и его расщепление. Справа от грудины может определяться нежный диастолический шум, обусловленный усиленным кровотоком по верхней полой вене. В легких выслушиваются влажные, мелкопузырчатые, застойные хрипы, участки бронхиального дыхания. Явления недостаточности кровообращения нарастают вслед за одышкой и в разгар клинической картины выражены резко (гепатомегалия, отеки, набухшие вены шеи, напряженный родничок).
На электрокардиограмме всегда фиксируется правограмма, гипертрофия и перегрузка правых отделов сердца.
Рентгенологическое исследование выявляет увеличение размеров сердечной тени, гипоплазию аорты и увеличение дуги легочной артерии. Легочный рисунок усилен. При впадении легочных вен в левую верхнюю полую можно наблюдать характерную тень сердца в виде цифры 8. Решающее значение для диагностики имеет рентгеноконтрастная ангиокардиография из правого желудочка или из легочной артерии с длительной съемкой, позволяющая проследить легочный кровоток и уровень впадения аномальных легочных вен в венозную систему большого круга кровообращения. Прогноз крайне тяжелый, около 75 — 80% детей умирают, не достигнув 1 года жизни.
Лечение начинают с дигитализации и противопневмонической терапии. После установления топического диагноза показана операция. Хирургическое вмешательство состоит в создании соустья между левым предсердием и коллектором, в который впадают легочные вены. Летальность при таких операциях остается высокой из-за частого развития недостаточности внешнего дыхания и левожелудочковой слабости. По-видимому, у новорожденных большего эффекта можно ожидать от паллиативной процедуры — атриосептостомии по Рашкинду, которая у таких больных разгружает малый круг кровообращения (Serrato е. а., 1968). Успешные операции при инфракардиальных формах порока пока носят характер единичных сообщений (Cooley, 1962; Jegeir е. а., 1967).
Двойная дуга аорты
Этот порок развития проявляется у новорожденных признаками респираторных нарушений в связи с ущемлением трахеи между дугами аорты и сдавлением ее просвета.
Эмбриогенез. У эмбрионов человека образуется шесть пар дуг аорты, связывающих вентральные или дорсальные аортальные стволы. Каждая из этих пар проходит в определенной бранхиальной (жаберной) дуге и имеет правую и левую ветви. В процессе эмбриогенеза I, II и V дуги аорты либо дегенерируют, либо трансформируются в соответствующие артериальные сосуды. Четвертая дуга слева сохраняется в виде аорты, а справа образует проксимальную часть подключичной артерии. При персистировании правой ветви. проходящей обычно позади пищевода, дуга аорты оказывается разделенной на два ствола, между которыми заключены трахея и пищевод. Если при сохранении правой ветви четвертой дуги аорты левая ветвь атрофируется и исчезает, возникает правосторонняя дуга аорты, ее передний (ассмановский) тип. Дуга аорты при этом проходит спереди трахеи слева направо и обычно сочетается с декстронозицией аорты или другими пороками сердца (тетрада Фалло, комплекс Эйзенменгера). Сдавление трахеи при этом виде аномалии возникает реже, чем при двойной дуге аорты. Последняя встречается в виде полного и неполного удвоения, когда правая ветвь четвертой эмбриональной дуги аорты не развивается в полную правостороннюю дугу аорты, а левая ветвь полностью не исчезает. От нее остается различных размеров рудиментарный дивертикул, от которого отходят левая подключичная артерия, артериальный проток или артериальная связка. В результате вокруг трахеи и пищевода создается кольцо, образуемое правосторонней дугой аорты, аортальным дивертикулом, артериальным протоком или его соединительнотканным рудиментом и легочной артерией. Эта аномалия известна как задний (аркиновский) тип правосторонней дуги аорты (В. Ионаш, 1960).
Клиническая картина. Симптоматика зависит от степени сдавления трахеи и пищевода и заключается в затрудненном прохождении пищи и нарушении дыхания. В большинстве случаев дыхание носит свистящий или стридорозный характер. Может отмечаться инспираторная одышка. Цианоз бывает временным или постоянным. Некоторые дети требуют непрерывной кислородотерапии. Новорожденные держат голову откинутой назад, так как в этом положении воздух легче проходит через сужение трахеи (Ст. Димитров, 1960). Нередко наблюдается упорный сухой кашель. При сдавлении возвратного нерва он приобретает металлический лающий оттенок, а голос ребенка становится хриплым. Кормление усугубляет симптоматику. Часто возникает срыгивание, рвота. Рвотные массы могут содержать свежую или измененную кровь — признак эзофагита. Вскоре развивается аспирационная пневмония, которая нередко приводит таких больных к гибели. Дисфагические явления могут быть менее выраженными в первый месяц жизни и усилиться при переходе на кормление твердой пищей. Больные подвержены респираторным заболеваниям. Присоединение ларинготрахеита значительно затрудняет дыхание и может вызвать тяжелую гипоксию.
Диагностику осуществляют путем рентгенологического и инструментального исследования. На обзорной рентгенограмме трахеи в профильной проекции можно обнаружить сужение ее просвета на уровне III — IV грудных позвонков. При исследовании пищевода с бариевой взвесью на этом же уровне определяют вдавление задней стенки пищевода. Ценные данные может дать томография. При трахеобронхоскопии просвет трахеи выглядит сдавленным в переднезаднем направлении из-за нависания передней стенки. Четко видна передаточная пульсация аорты. Необходимости в контрастном исследовании трахеи обычно не возникает. Ангиография позволяет определить расположение дуги аорты, ее ветвей и размеры стволов в случае двойной дуги.
Лечение. Показания к хирургическому лечению возникают у детей с тяжелыми нарушениями дыхания и дисфагией. При консервативном лечении подобные больные доживают лишь до 1 — 2-летнего возраста. Смерть может наступить внезапно или явиться следствием повторных пневмоний. В самых тяжелых случаях операцию производят в период новорожденности. При этом приходится выбирать между риском хирургического вмешательства и степенью выраженности симптомов, главным образом дыхательной недостаточности.
Хирургическое вмешательство заключается в перевязке и пересечении более узкой дуги, обычно передней. При неполном удвоении дуги аорты производят перевязку артериального протока или артериальной связки, сдавливающей трахею. Если причиной дыхательных нарушений является сдавление трахеи правосторонней дугой аорты в результате смещения ее влево ветвью подключичной артерии, возможна перевязка и рассечение последней, так как кровоснабжение плеча в достаточной мере обеспечивается анастомозами (В. Ионаш, 1960).