ХИРУРГИЯ НОВОРОЖДЕННЫХ - 1976
2. ЧАСТНЫЕ РАЗДЕЛЫ
7. Генетически обусловленные заболевания и пороки развития
Изучение наследственно обусловленных пороков развития основано на достижениях молекулярной биологии, цитологии, медицинской генетики, клинической биохимии и др. Большой вклад в развитие медицинской п клинической генетики в нашей стране внесли Н. П. Дубинин, В. Д. Тимаков, Н. И. Шуков-Вережников, II. II. Бочков, Е. Ф. Давиденкова, А. А. Прокофьева-Бельговская, Б. В. Конюхов и др.
Под влиянием неблагоприятных генетических факторов любые органы и ткани новорожденного ребенка могут оказаться аномально измененными. При этом в одних случаях врожденный порок касается лишь отдельного органа, в других он является составной частью сложного клинического синдрома, в который входят аномалии конечностей, глаз, лицевого скелета, внутренних органов. Развитие подобных множественных пороков нередко сочетается с нарушением обмена веществ, нейрогуморальных и нейроэндокринных процессов.
Биологические подходы к проблеме врожденных пороков основываются на учете определенных уровней организации наследственных структур. В настоящее время принято выделять три таких уровня: 1) генный (молекулярный) уровень; 2) наследственный аппарат хромосом (хромосомный уровень); 3) систему совокупности хромосом (геномный уровень). Мутации, происходящие на одном из трех уровней, являются причиной наследственных болезней и врожденных пороков развития.
Клиническая картина наследственных аномалий часто неотличима от аномалий экзогенного происхождения (проблема фенокопирования). Это можно объяснить тем, что в обоих случаях в эмбриогенезе изменяются одни и те же физиологические и биохимические процессы в первом случае под влиянием недостаточно генетически детерминированных ферментных систем, во втором — под влиянием факторов внешней среды. Аномалии плода в период его внутриутробного развития тесно связаны с состоянием организма матери. Клинические наблюдения показывают, что частота врожденных аномалий возрастает по мере увеличения возраста матери. Возможность повреждения эмбриона и плода особенно велика в так называемые критические периоды его развития, т. е. в моменты эмбриогенеза, когда процессы дифференциации тех или иных структур протекают особенно интенсивно.
Все врожденные пороки развития, связанные с генетическими факторами, можно условно разделить на три группы: 1) обусловленные генными мутациями; 2) обусловленные хромосомными мутациями; 3) частично обусловленные генетическими факторами.
Пороки развития, обусловленные генными мутациями
Наследственный характер заболевания подразумевает прежде всего его семейное распространение. Однако большинство пороков развития встречается не в виде семейных, а в виде спорадических (изолированных) случаев. Поэтому деление наследственных аномалий на семейные и спорадические сугубо условно, а их противопоставление при клинико-генетическом изучении не оправдано.
Общая характеристика клинических проявлений наследственного порока зависит от действия одного или нескольких патологических (мутантных) генов. В этом отношении действие патологического гена может быть охарактеризовано его пенетрантностью (частотой проявления) и экспрессивностью (степенью развития контролируемого им признака).
Клинико-генетический анализ требует тщательного изучения не только типичных, но и атипичных, абортивных и стертых случаев данной аномалии. В связи с клиническим полиморфизмом, столь характерным для наследственных заболеваний, важное значение приобретает выявление как типичных симптомов, так и микросимптомов данного заболевания.
Наследственные формы заболеваний характеризуются определенным типом наследственной передачи. Chung с соавт. (1960) предложили критерии для определения основных типов наследования: аутосомно-доминантного, аутосомно-рецессивного, наследования через Х-хромосому (сцепленного с полом).
Аутосомно-доминантное наследование с полной пенетрантностью характеризуется следующими признаками: 1) прямая передача из поколения в поколение «без скачков», связанных с пропуском поколений; 2) у каждого больного ребенка один из родителей болен; 3) в браке больного — гетерозиготы со здоровым партнером ожидаемая частота появления больных детей 50%; 4) оба пола поражаются в равной степени; 5) «доминантные» гены известны только по их клинической характеристике у гетерозигот; гомозиготы встречаются исключительно редко.
Если носители патологического доминантного гена клинически здоровы (так называемая неполная пенетрантность), то заболевание «перескакивает» одно или несколько поколений и частота появления больных детей снижается. У больных со стертой (абортивной) формой заболевания поставить диагноз можно лишь после тщательного клинико-генетического и лабораторного обследования (рис. 28).
Рис. 28. Аутосомно-доминантный тип наследования при множественных хрящевых экзостозах в трех поколениях (собственное наблюдение).
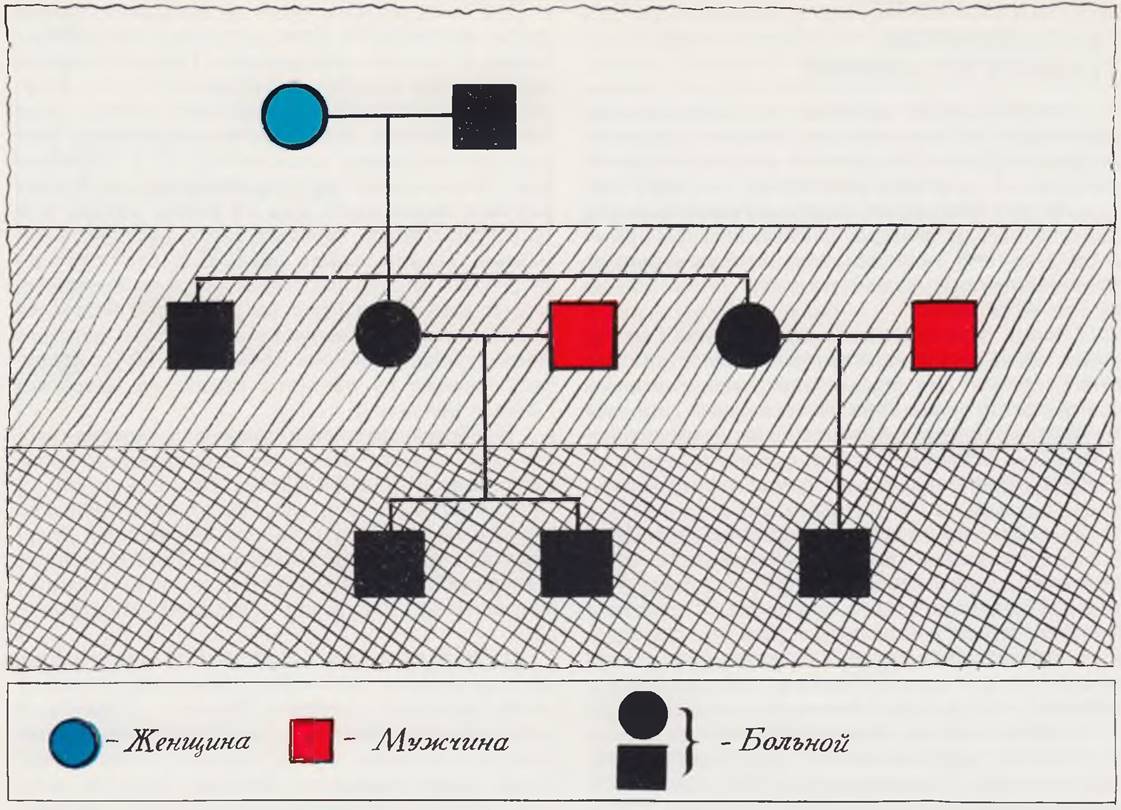
При доминантном типе наследования крайне редко наблюдается брак двух лиц, страдающих одним и тем же заболеванием. Больной практически всегда является гетерозиготой, т. е. носителем нормального и аномального генов, тогда как клинически здоровые лица в данной семье здоровы не только клинически, но и генетически. Отягощенная наследственность в подобных случаях имеет место или по линии матери, или по линии отца, т. е. носит «односторонний» характер. Теоретический риск заболевания каждого ребенка можно определить еще до первой беременности. Если один из родителей болен, то возможность заболевания первого и каждого последующего ребенка составляет 50 %. Здоровый ребенок не является носителем патологического признака.
Наиболее типичным примером доминантного наследования являются синдактилии, полидактилии и полифалангии, макродактилии и брахидактилии. Эти пороки нецелесообразно устранять брачным отбором, ибо они не представляют жизненной опасности. Перечисленные аномалии нередко входят в состав сложных клинических синдромов (синдром Марфана и др.).
Другим примером системного заболевания, обусловленного дегенеративными изменениями кожи, является синдром Элерса — Данлоса с аутосомно-доминантным, или Х-сцепленным, типом наследования. Среди пороков развития, которые могут привлечь внимание хирурга, следует назвать диафрагмальную грыжу, эктазию частей пищевода и дыхательного тракта, расслаивающие аневризмы аорты.
По аутосомно-доминантному типу наследуются такие пороки, как коарктация аорты, дефект межпредсердной перегородки, открытый артериальный проток, артрогриппоз (синдром множественных врожденных контрактур), синдром Турнера — Кизера (гипоплазия коленной чашечки, вывих коленного сустава и др.), синдром Нейвергелта, связанный с пороком развития верхних и нижних конечностей, нейрофиброматоз (болезнь Реклингаузена), полипоз желудочно-кишечного тракта, ретинобластома, врожденная лимфедема, туберозный склероз (болезнь Бурневилля), различные дизостозы (ключично-черепной, челюстно-лицевой, черепно-лицевой), несвоевременный остеогенез, наследственный птоз, эктопия хрусталика и др.
Помимо доминантного типа передачи наследственности, определяемой одной парой генов (моногибридная наследственность), возможна ди- и полигибридная передача, сцепленная или связанная с полом и ограниченная полом.
Аутосомно-рецессивное наследование с полной пенетрантностью характеризуется следующими признаками: 1) родители и родственники, кроме сибсов (братьев-сестер), обычно здоровы; 2) если рецессивные патологические гены являются аллельными, т. е. расположенными в одинаковых участках гомологичных хромосом, то все дети двух больных родителей будут больны; 3) от брака двух клинически здоровых гетерозигот 25% детей будут больными и 50% — здоровыми носителями, подобно своим родителям; 4) оба пола поражаются в равной степени; 5) если заболевание редкое, то повышается значение кровного родства родителей.
Неполная пенетрантность при аутосомно-рецессивном наследовании является серьезной проблемой при генетическом анализе, так как в этих семьях больных может оказаться больше, чем предсказывают теоретические расчеты (рис. 29).
Рис. 29. Аутосомно-рецессивный тип наследования при врожденной миопатии.
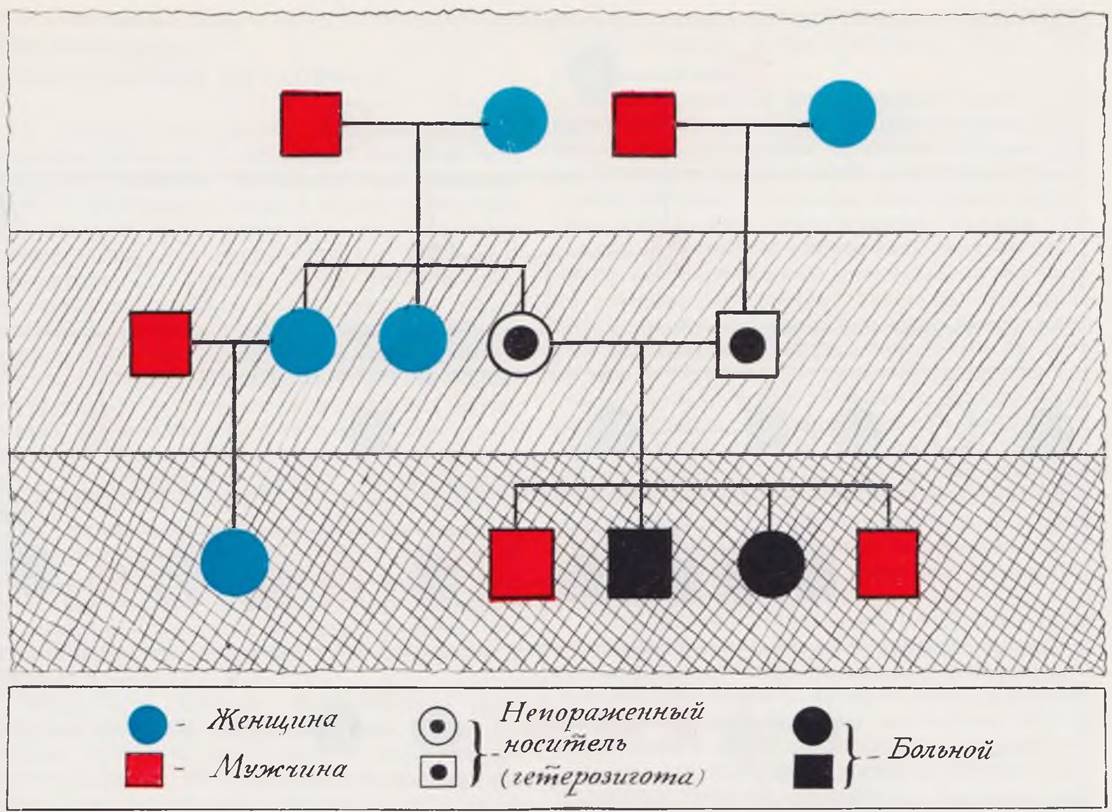
Несмотря на то что среди рецессивных заболеваний встречаются преимущественно различные энзимопатии (так называемые метаболические блоки), пороки развития также нередко имеют рецессивный тип наследственной передачи. К ним относят акроцефалосиндактилию (синдром Апера), ахейроподию (отсутствие кистей п стоп), рецессивные формы артрогрппоза, хондроэктодермальную дисплазию (синдром Эллиса — Ван-Кревельда), умственную отсталость и пигментный ретинит в сочетании с гипогонадизмом, тучностью и полидактилией (синдром Лоренса — Муна — Бидля), содружественное врожденное косоглазие, гаргоилизм (мукополисахаридозы), некоторые формы полипов кишечника, из которых в последующем могут развиваться злокачественные новообразования, и др.
Кроме того, имеется обширная группа врожденных пороков, которую лишь условно можно отнести к рецессивному типу наследования, так как наблюдаются преимущественно спорадические формы. К ним относят отсутствие различных мышечных групп (шеи, грудной клетки и др.), косорукость, атрезии пищевода и различных отделов кишечника, аномалии поджелудочной железы (кольцевидная, кистофиброзная), эмбриональные и диафрагмальные грыжи, спинномозговые грыжи, эктопию мочевого пузыря. Так как тип наследственной передачи в этих случаях окончательно не установлен, мы можем лишь условно рассматривать их как рецессивные.
Рецессивные наследственные пороки клинически выявляются в тех случаях, когда оба родителя оказываются носителями рецессивного гена (так называемыми гетерозиготами). По теоретическим расчетам вероятность заболевания составляет 25% (гомозиготы по рецессивному патологическому гену), возможность рождения гетерозиготы — 50 %, здорового ребенка (гомозиготы нормального гена) — 25%. Брак больного со здоровым будет давать в потомстве 100% гетерозигот. Брак больного с гетерозиготой будет давать в потомстве 50% больных и 50% гетерозигот (носителей). Выявление гетерозигот приобретает особое значение в случаях родственных браков в семьях, где имеется данная рецессивная аномалия. Клиническим доказательством гетерозиготности могут быть «малые» признаки заболевания. Однако нередко ни клинические, ни клинико-лабораторные методы не позволяют с достоверностью выявить скрытых носителей патологического гена, что крайне осложняет заключение о наследственном характере данной аномалии. Следует учитывать, что, кроме простой аутосомно-моногибридной рецессивной наследственности, зависящей от одной пары рецессивных признаков, имеются и другие ее формы: полигенная рецессивная наследственность; рецессивная, связанная с полом наследственность, рецессивная, ограниченная полом наследственность.
При наследовании через Х-хромосому (наследование, сцепленное с полом) отмечается следующее: 1) родители и родственники, за исключением родственников мужского пола по материнской линии, обычно здоровы; 2) гомозиготные больные мужчины не передают заболевания своим детям, но все их дочери являются гетерозиготными носителями; 3) гетерозиготные женщины-носители клинически здоровы, но передают заболевание 50% своих сыновей, а 50% их дочерей являются гетерозиготами, подобно своим матерям; 4) больные женщины рождаются только от брака женщин-носителей и больных мужчин; 5) каждый больной мужчина рождается от женщины-носителя. Кроме того, возможно возникновение неомутаций.
Когда патологический ген локализован в Х-хромосоме, то в зависимости от того, является он доминантным или рецессивным, выделяют доминантную, сцепленную с полом наследственность и рецессивную, сцепленную с полом наследственность (рис. 30). От сцепленной с полом наследственности следует отличать наследственность, ограниченную полом. Типичным примером полностью ограниченного полом доминантного признака является гипоспадия.
Рис. 30. Сцепленное с полом наследование врожденных аномалий конечностей.
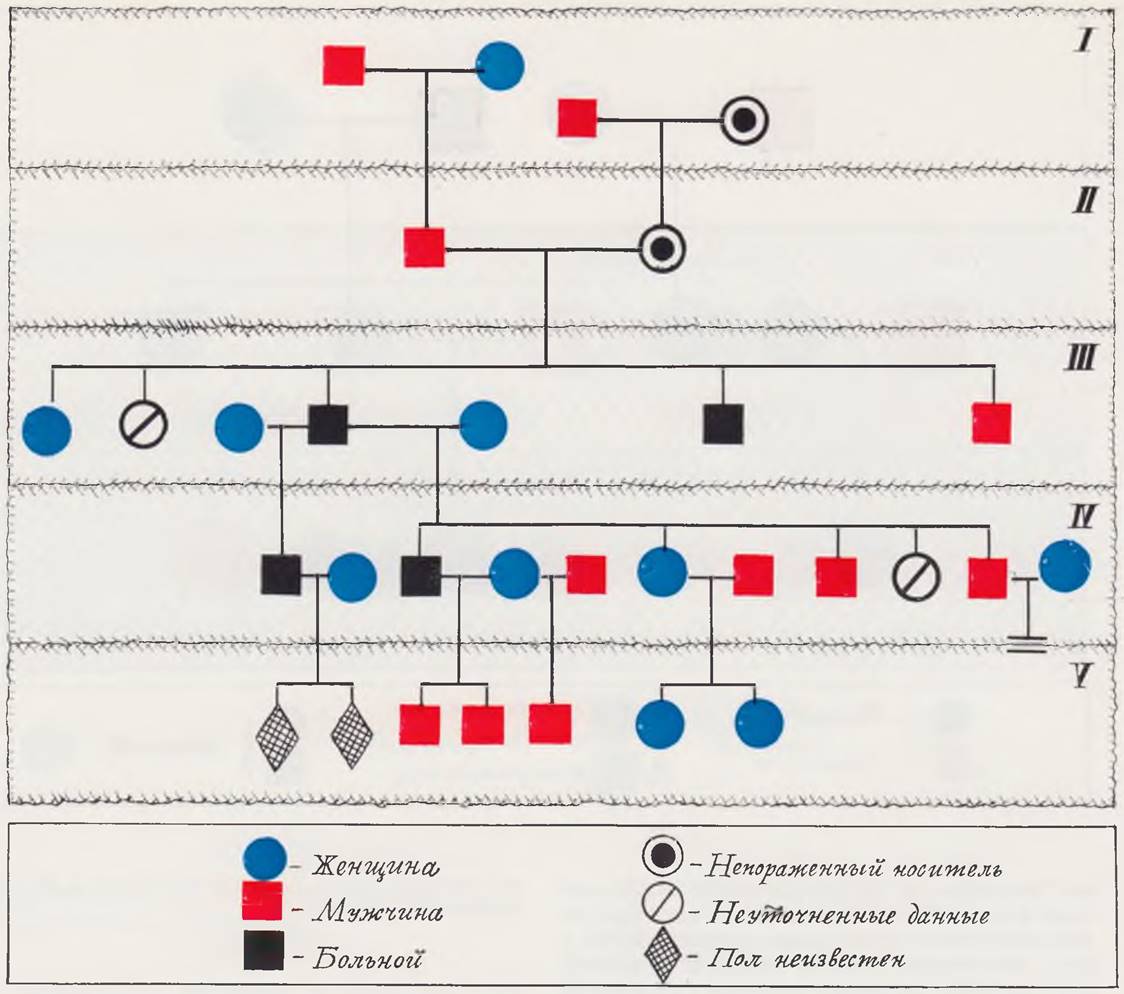
Пороки развития, обусловленные хромосомные мутациями
По данным ВОЗ (1965), генетический риск рождения ребенка с серьезным пороком или аномалией развития составляет как минимум 2,5%. В последующие годы в процессе широких исследований было показано, что тяжелыми врожденными аномалиями страдает около 5% новорожденных (Jacobs е. а., 1970, и др.). Врожденные аномалии ассоциируются с отклонениями кариотипа у 70,2% новорожденных, причем более половины их рассматриваются как лица с нормальными вариациями признаков. Значительное количество соматических врожденных пороков связано с числовыми или структурными изменениями хромосом (аберрациями). В популяции синдром Клайнфелтера встречается с частотой 0,25 %, синдром Дауна — 0,2%, синдром трипло-Х — 0,125%, синдром Шерешевского — Тернера — 0,04 %, синдром Эдвардса — 0,02%, синдром Патау — 0,007%. Среди 22 пар аутосом и пары половых хромосом, составляющих кариотип человека, наиболее частыми являются аберрации 13-й, 18-й, 21-й и половых хромосом X и Y.
Нарушения развития, обусловленные аберрациями хромосом, можно разделить па две категории: соматические пороки и аномалии полового развития. Большая часть нарушений полового развития связана с числовыми или структурными аберрациями половых хромосом. Однако при аберрациях аутосом дефекты формирования половых органов не столь уж редки, а при аберрациях половых хромосом соматические дефекты являются скорее правилом, чем исключением.
При аберрациях аутосом большинство симптомов не являются специфичными для данного конкретного синдрома. Например, синдактилия наблюдается при болезни Дауна и трисомии по 18-й хромосоме, аномальные по форме низко посаженные ушные раковины — при синдроме «кошачьего крика», трисомиях по 13-й и 18 й хромосомам, при синдроме 18 q (делеция1 длинного плеча 18-й хромосомы) и при синдроме 18 р (делеция короткого плеча 18-й хромосомы). Врожденные расщелины верхней губы встречаются при трисомиях по 13-й и 18-й хромосомам, сращение верхнего и нижнего века (эпикант) — при трисомиях по 18-й и 21-й хромосомам и при синдромах 18 q — и 18 р —. При синдроме «кошачьего крика» и синдромах 18 р — и 18 q — имеется гипертелоризм (широкое расстояние между глазами), а при синдромах Патау, Эдвардса — грыжи.
1 Делеция — структурное изменение хромосомы, связанное с утратой части ее.
Пороки развития опорно-двигательной и соединительнотканной систем. При наличии добавочной 18-й хромосомы (трисомия-18) внешний вид больного весьма характерен. Череп удлиненной формы, с широким зависающим затылком, узким лбом, недоразвитой нижней челюстью, врожденной расщелиной верхней губы, низко расположенными аномальными по форме ушными раковинами, маленьким треугольным ртом, птозом. Грудная клетка как бы сжата в вертикальном направлении: бочкообразная по форме, короткая, с широкой грудиной. Таз обычно узкий, бедра сближены, подвижность тазобедренных суставов сильно ограничена. Типичны аномалии стоп (talipes equinovarus) и кистой: ладонь широкая с короткими пальцами и особым положением II и V пальцев в связи с гипоплазией мышц тенара и гипотепара. В ряде случаев у больных имеются синдактилия, аномалии ногтей, паховые н пупочные грыжи, помутнение роговицы.
Для больных с трисомией по 21-й хромосоме характерен маленький круглый череп со сглаженным плоским затылком, у них недоразвита верхняя челюсть, круглое лицо с коротким широким носом, запавшим переносьем, близко посаженными раскосыми глазами (направление разреза снаружи и сверху вниз и внутрь), аномальными ушными раковинами. Рот часто полуоткрыт, зубы аномальные по форме, кариозные, язык толстый с поперечными бороздами. Рост обычно небольшой, конечности укорочены, часто наблюдается повышенная подвижность суставов из-за гипотонии мышц, недоразвития связок или суставных впадин, возможно наличие грыж, синдактилий между II и III пальцами стоп, увеличенного расстояния между I и II пальцами ног, искривленных и укороченных мизинцев рук, поперечной борозды на ладони.
При трисомии по 13-й хромосоме наблюдаются брахицефалическое строение черепа, микроцефалия, врожденные расщелины верхней губы и неба, микрофтальмия или анофтальмня, уродливые низко расположенные уши, недоразвитая нижняя челюсть, помутнение роговицы. Нередки пороки развития ступней, повышенная гибкость суставов больших пальцев, ульпарная полидактилия и синдактилия, грыжи, колобома.
Синдром «кошачьего крика» обусловлен отсутствием части короткого плеча хромосомы 5, что выражается в делении половины короткого плеча или в наличии кольцевой хромосомы, или в наличии сбалансированной транслокации 5-й хромосомы и какой-либо другой хромосомы. Из-за аномального строения гортани тембр голоса напоминает кошачье мяуканье. Этот симптом и послужил основанием для названия синдрома. Обычными симптомами являются микроцефалия, круглое лицо с близко посаженными раскосыми глазами, эпикант, низко расположенные аномальные по форме ушные раковины, микро- и ретрогнатия, мышечная гипотония, парезы конечностей.
Соматические аномалии синдрома делеции длинного плеча хромосомы 18 выражаются в микроцефалии, диспропорции лица (уменьшение средней его части, аномальные низко расположенные уши с резко выдающимся антитрагусом, аномалиях среднего уха, гипертелоризме, эпиканте, мышечной гипотонии.
Пороки развития внутренних органов и центральной нервной системы. При синдроме 18 q — (делеция длинного плеча хромосомы 18) наблюдаются пороки развития почек, сердца. При синдроме Дауна отмечаются дисфункции желез внутренней секреции с соответствующими изменениями. Изменения со стороны мозга заключаются в гипоплазии мозжечка и ствола мозга, водянке желудочков мозга, различных морфологических аномалиях нейронов. Среди пороков сердца отмечаются незаращение артериального протока, межжелудочковой пли межпредсердной перегородки, коарктация аорты, тетрада Фалло. При синдроме трисомии по 18-й хромосоме уже при рождении можно обнаружить маленькую плаценту, единственную пупочную артерию. Пороки развития почек обычно выражаются в наличии двойного мочеточника, гидронефроза, пороки сердца — в незаращении артериального протока или межжелудочковой перегородки. Со стороны мозга отмечается демиелинизация проводящих путей больших полушарий и мозжечка. При трисомии по 13-й хромосоме регистрируются пороки развития ночек, сердца, мозга, отмечают гемангиомы лица. Из пороков сердца чаще выявляются незаращение артериального протока п межжелудочковой перегородки. Аномалии мозга выражаются в уменьшении количества извилин, смещении отделов мозга, анэнцефалии.
Соматические аномалии при аберрациях половых хромосом наиболее выражены при моносомии по Х-хромосоме (синдром Шерешевского — Тернера). Больные с полной или частичной моносомией отличаются небольшим ростом, имеют укороченную шею с низким уровнем оволосения на затылке. Пороки развития наблюдаются со стороны мочевыделительной, сердечно-сосудистой, костно-сосудистой, костно-мышечной и соединительнотканной системы. Часто наблюдается диспозиция и диспропорция мозгового и лицевого черепа. Характерны множественные аномалии скелета, задержка окостенения, отсутствие слияния эпифизов с метафизами, остеопорозы, раннее прорезывание и деформация зубов вследствие идиопатической резорбции корней, недоразвитие нижней челюсти (микрогнатия пли ретрогнатия), аномалии прикуса. Часто встречаются деформации позвоночника (кифоз, сколиоз, spina bifida, сращение позвонков, сращение ребер, укорочение тела позвонка). Со стороны конечностей часты деформации лучезапястных суставов, вальгусное положение стоп, вальгусная девиация локтевых н коленных суставов, синдактилия. Укорочение четырех метакарпальных и метатарзальных костей приводит к укорочению четырех пальцев кистей и стопы, иногда наблюдаются укорочение и искривление других пальцев, синдактилия. Со стороны мышечной системы выявляется отсутствие или гипоплазия отдельных глазных мышц, что, возможно, является причиной косоглазия и астигматизма. Отсутствие леватора верхнего века ведет к птозу. Следует отметить слабость связочного аппарата суставов, деформацию ногтей, наличие морфологически аномальных ушных раковин, боковых крыловидных складок на шее. При синдроме Шерешевского — Тернера нередко обнаруживаются коарктация аорты, транспозиция крупных сосудов, незаращение артериального протока, стенозы устья аорты или легочной артерии. Нередко наблюдаются аномалии почек, выражающиеся в дисплазии одной из них или аномальном подковообразном строении, удвоении мочеточников или лоханок.
Пороки развития внутренних органов при полисомиях выражены значительно слабее и не столь часты, как при моносомиях. Фенотип больных с генотипом XYY характеризуется высоким ростом, макрогнатией, а аномалий часто вообще не выявляется.
При синдроме Клайнфелтера при генотипе XXY аномалии развития выявляются в различном возрасте, начиная с периода новорожденности. При увеличении количества добавочных хромосом частота аномалий возрастает: встречается вальгусная девиация и излишняя подвижность суставов, радио-ульнарный синостоз. У больных обычно высокий рост, удлиненные конечности, евнухоидное телосложение, склонность к ожирению.
При отклонении числа обеих половых хромосом, X и Y, в сторону их увеличения (полисомии), обычно наблюдаются черты акромегализма, аномалии со стороны сердечно-сосудистой системы.
Механизмом, обеспечивающим числовые аберрации хромосомы типа моносомий, трисомий и полисомий, считается нерасхождение хромосом, хотя этиология и механизмы, ведущие к нерасхождению, изучены пока недостаточно. Можно, однако, полагать, что и в данном случае решающую роль играют реальные клеточные механизмы, обусловливающие пространственную структурную организацию хромосом клеточного ядра. В результате нерасхождения в процессе первого и второго деления меноза могут возникнуть следующие типы гамет: X, Y, О, XY, XX, XY, XXY, XYY, XXYY, X, О, XX, XXX, ХХХХ. Зиготы, имеющие комплекс половых хромосом YY, YO, ОО, являются летальными, а большая часть хромосомных аномалий, обусловленных слиянием нормальных и аномальных зигот, обнаружена и исследована.
Наличие у пациентов нескольких генотипически различных хромосомных линий (клонов) называют мозанцизмом. Процентное соотношение клонов, если не учитывать дальнейшее селективное преимущество одного из них, будет зависеть от стадии, в которой произошло нерасхождение. Клиническая картина зависит от преобладания того или иного клеточного клона в соответствующей ткани или органе и от взаимодействия функционирующих генов в клетках при аномальном хромосомном комплексе. При различных структурных изменениях одной из Х-хромосом, связанных с частичной моносомией, клиническая картина синдрома Шерешевского — Тернера несколько сглаживается. Четких клинических различий различных форм синдрома пет. Весьма разнообразны кариотипические варианты так называемой, группы смешанной дисгенезии гонад, обусловленные мозаичностью кариотипа, например, 45, Х/46, XX; 45, Х/46, XX р 45 Х/46, XXqi; 45, Х/46, XY; 45, ХО/46, ХХ/46, XY и т. д. Наличие клеточного клопа 46, XX обусловливает развитие овариальной ткани и, следовательно, возможность овуляции, менструирования и деторождения. Клоны клеток 46, XY или клон с делетированной Y-хромосомой обусловливают наличие тестикулярной ткани, а это ведет к верилизации, мужскому типу телосложения, увеличению клитора или даже развитию почти нормального по величине penis.
Соматические аномалии встречаются реже. Иногда этот клинический вариант называют смешанной гонадальной дисгенезией. Довольно редко встречается дисгенезия гонад при кариотипе 46, XY, т. е. кариотип и фенотип не соответствуют друг другу.
При этой форме вторичные половые признаки обычно не выражены, соматические аномалии отсутствуют, а наружные и внутренние половые органы недоразвиты. Индивидуумы с мужским псевдогермафродитизмом имеют кариотип 46, XY и очень редко 45, Х/46, XYq —; 45, Х/47, XXq — у; 45, ХО/46, XY и т. п. Всех больных
можно разделить на две подгруппы: 1) индивидуумы с двойственными или преимущественно мужскими наружными гениталиями; 2) индивидуумы с женскими наружными гениталиями и развитыми молочными железами. При истинном гермафродитизме около 60% гермафродитов имели кариотип 46, XX и у 40% в кариотипе имелась нормальная или делетированная Y-хромосома. Однако роль хромосом в данных случаях выяснена недостаточно ввиду невозможности отрицания, скрытого мозаицизма или транслокации фрагмента Y-хромосомы на одну из аутосом.
В подавляющем большинстве случаев при дисгенезии семенных канальцев клиницисты имеют дело с синдромом Клайнфелтера, обусловленным полисомией по X-хромосомам.
Врожденные аномалии полового развития встречаются не только при аберрациях половых хромосом, но и при числовых и структурных аномалиях аутосом. Так, при синдромах трисомии по 13-й и 18-й хромосомам наблюдается крипторхизм, при синдроме трисомии по 21-й хромосоме — гипоплазия половых органов (недоразвитие их, видимо, связано с дисфункцией половых желез, наблюдаемой обычно у дошкольников). После 12 лет половые органы у мальчиков могут достигнуть нормальных размеров.
Пороки развития, частично обусловленные генетическими факторами
К этой группе относят врожденные пороки сердца, пилоростеноз, незаращение губы и неба, анэнцефалию и spina bifida, врожденный вывих бедра, косолапость с конской стопой. Вероятность этих пороков определяется исключительно эмпирически. «Эмпирический риск» означает вероятность появления данного порока, основанную на практике и наблюдениях, а не на теоретических расчетах. У новорожденных эти пороки развития наблюдаются с частотой 1 — 3 на 1000.
Генетическая гетерогенность этих пороков проявляется в том, что в одних семьях они встречаются чаще, в других реже. На значение генетических факторов в их происхождении указывают более высокая распространенность дефекта среди больных родственников, чем среди данной группы населения, непостоянная частота у обоих полов, сравнительно высокая степень конкордантности между однояйцевыми близнецами.
Пилоростеноз — относительно частое заболевание первых месяцев жизни. Отмечена связь между частотой случаев и кровным родством родителей, а также более высокая конкордантность между однояйцевыми близнецами, чем между двуяйцевыми. Так, частота заболевания второго близнеца в случае заболевания первого для однояйцевых близнецов составляет 67%, тогда как для двуяйцевых — 3 %. Наиболее убедительные данные получают при анализе наследственно-семейных случаев. Заболеваемость оказывается выше всего среди лиц мужского пола — родственников больной женщины и ниже всего — среди родственников больного мужчины.
Врожденные незаращения губы, неба н альвеолярного отростка с позиций эмбриогенеза иногда рассматривают как различные степени одного и того же дефекта. Клинико-генетический анализ показывает, что среди родственников больного (пробанда) с расщелиной губы или с сочетанием расщелины губы и неба часто наблюдаются больные с расщелиной губы и неба, тогда как повышения частоты изолированной расщелины неба не отмечается. Вместе с тем среди родственников пробанда с расщелиной неба наблюдается увеличение числа лиц с этой аномалией, но без увеличения числа лиц с расщелиной неба в сочетании с расщелиной губы. И хотя могут существовать исключения, можно считать, что одни и те же пороки развития имеют различную наследственную этиологию. На роль генетических факторов указывают также нередко выраженный семейный характер и боле высокая конкордантность у однояйцевых близнецов, чем у двуяйцевых. Так, частота заболеваний второго близнеца в случае заболевания первого для однояйцевых близнецов составляет 33 %, тогда как для двуяйцевых — лишь 5%.
Клинико-генетический анализ показывает, что расщелины губы и неба чаще встречаются среди близких, чем среди дальних родственников пробанда. Так, если среди населения расщелина губы составляет менее 0,1%, а неба — 0,04%, то в семьях, где уже имеется больной ребенок при здоровых родителях, вероятность заболевания следующего ребенка возрастает до 4% для расщелины губы и до 1,8% для расщелины неба. Если же имеется больной ребенок, а один из родителей также имеет эту аномалию, то вероятность заболевания следующего ребенка еще более возрастает — до 14 — 17%.
Анэнцефалия и spina bifida могут носить семейный характер и особенно часты среди сибсов в семьях, где этой аномалией страдает уже один ребенок. Зависимость частоты случаев от кровного родства родителей подтверждает роль генетических факторов. В настоящее время трудно выдвинуть гипотезу образования этих аномалий, основанную лишь на каком-либо одном факторе (наследственном или экзогенном). По-видимому, об анэнцефалии можно говорить скорее, как о полиэтиологическом синдроме.
Врожденный вывих бедра. Клинико-генетический анализ показывает более высокую конкордантность однояйцевых близнецов, чем двуяйцевых. Так, частота заболевания второго близнеца в случае заболевания первого для однояйцевых близнецов составляет 41,4%, а для двуяйцевых — 2,8 %. Имеются также указания на более широкую распространенность этой аномалии среди сибсов и потомков больных лиц. Если среди населения врожденный вывих бедра составляет 0,1%, то в тех семьях, где уже имеется один больной ребенок, вероятность заболевания второго возрастает при здоровых родителях до 5 %, а в случае если один из родителей имеет эту же аномалию — до 10 — 15 %.
Косолапость и конская стопа наиболее часто встречаются среди родственников пробанда, чем среди здорового населения. Если в популяции этот порок составляет 0,1%, то в семьях, где уже имеется один больной ребенок, вероятность заболевания второго возрастает при здоровых родителях до 5 — 10%. При исследовании близнецов наблюдается более высокая конкордантность однояйцевых близнецов, чем двуяйцевых. Так, частота заболевания второго близнеца в случае заболевания первого для однояйцевых близнецов составляет 32%, а для двуяйцевых — 3%.
Изучение генетически обусловленных врожденных пороков развития имеет значение не только для своевременной диагностики и лечения, но и для генетического прогноза. Медико-генетическое консультирование при врожденных пороках развития ограничивается наиболее важными сторонами профилактики: предоставление родителям объективной информации о возможном риске заболевания детей; разъяснение последствий браков между гетерозиготами — носителями одного и того же мутантного гена (особенно при кровном родстве); правильные советы семьям, где уже имеются врожденные аномалии развития (о нежелательности рождаемости у матерей пожилого возраста и др.). В подобных случаях следует прежде всего учитывать клинико-генеалогические и другие сведения о данной конкретной семье, в которой может быть ряд особенностей в типе наследственной передачи, пенетрантности, особенностях самого врожденного синдрома.
Задачи профилактики врожденных пороков развития требуют дальнейшего развития специализированных медико-генетических отделений и комплектования их высококвалифицированными кадрами генетиков-клиницистов.