Основы биоинформатики - Огурцов А.Н. 2013
Основания биоинформатики
Предмет биоинформатики
Особенность биоинформационных данных
Биологию традиционно считают описательной, а не аналитической наукой. Несмотря на то, что последние успехи науки не изменили это основное направление, радикально изменилась сущность биологических данных.
До последнего времени все биологические наблюдения носили в основном случайный характер, правда, с различным уровнем точности и некоторые были проведены действительно с очень хорошим качеством.
Первая особенность биологических данных последнего поколения исследований состоит в том, что данные стали не только количественными и более точными, но, как в случае нуклеотидных и аминокислотных последовательностей, они стали дискретными.
Расшифровать геномную последовательность индивидуального организма или клона стало возможным не только полностью, но и, что принципиально, точно. Ошибки эксперимента никогда не могут быть полностью исключены, но для современного секвенирования генома они чрезвычайно низки.
Это не означает, что биология стала аналитической наукой. Жизнь действительно подчиняется законам физики и химии, но она слишком сложна и зависима от цепи исторических случайностей, чтобы сегодня можно было бы детально объяснить её свойства, исходя из фундаментальных принципов. А достигнутая точность фиксации геномов не является достаточным условием для объяснения явления жизни.
Вторая очевидная особенность биоинформационных данных — это их огромное количество. Сейчас банки данных нуклеотидных последовательностей содержат около 20 млрд, нуклеиновых пар оснований. Если мы возьмем в качестве единицы измерения размер генома человека (НUmаn Genome Equivalent, HUGE), то этот объём информации эквивалентен 7 HUGE. База данных только белковых структур содержит более 86 000 записей, каждая из которых является полным описанием координат ~400 аминокислотных остатков данного белка в трёхмерном пространстве (рисунок 1) - http://www.pdb.org/.
Рисунок 1 - Веб-страница Банка белковых данных PDB
Огромны не только размеры отдельных банков данных, но и экспоненциальные темпы их увеличения. Так, например, в таблице 1 представлена динамика заполнения базы данных генетических последовательностей GenBank, http://www.ncbi.nlm.nih.gov/genbank/. А на рисунке 2 эти данные представлены в графическом виде.
Такое количество и качество биологических данных стимулирует исследователей к достижению следующих целей:
✵ Увидеть картину мира живых существ чётко и целиком, то есть понять интегрирующие аспекты биологии организмов, рассматриваемых как согласованные комплексные системы.
✵ Связать между собой последовательность, трёхмерную структуру, взаимодействия и функции отдельных белков, нуклеиновых кислот и их комплексов.
✵ Использовать данные о современных организмах как основу для изучения организмов во времени'.
- назад в прошлое, чтобы вычислить последовательность событий в эволюционной истории (филогенетический анализ),
- вперёд к научно обоснованной модификации биологических систем (биотехнология).
✵ Способствовать применению этих знаний в медицине, сельском хозяйстве и других областях.
Таблица 1 - Динамика роста базы данных GenBank
|
Год |
Число пар оснований |
Число последовательностей |
Год |
Число пар оснований |
Число последовательностей |
|
1982 |
680 338 |
606 |
1996 |
651 972 984 |
1 021 211 |
|
1983 |
2 274 029 |
2 427 |
1997 |
1 160 300 687 |
1 765 847 |
|
1984 |
3 368 765 |
4 175 |
1998 |
2 008 761 784 |
2 837 897 |
|
1985 |
5 204 420 |
5 700 |
1999 |
3 841 163 011 |
4 864 570 |
|
1986 |
9 615 371 |
9 978 |
2000 |
11 101 066 288 |
10 106 023 |
|
1987 |
15 514 776 |
14 584 |
2001 |
15 849 921 438 |
14 976 310 |
|
1988 |
23 800 000 |
20 579 |
2002 |
28 507 990 166 |
22 318 883 |
|
1989 |
34 762 585 |
28 791 |
2003 |
36 553 368 485 |
30 968 418 |
|
1990 |
49 179 285 |
39 533 |
2004 |
44 575 745 176 |
40 604 319 |
|
1991 |
71 947 426 |
55 627 |
2005 |
56 037 734 462 |
52 016 762 |
|
1993 |
157 152 442 |
143 492 |
2006 |
69 019 290 705 |
64 893 747 |
|
1994 |
217 102 462 |
215 273 |
2007 |
83 874 179 730 |
80 388 382 |
|
1995 |
384 939 485 |
555 694 |
2008 |
99 116 431 942 |
98 868 465 |
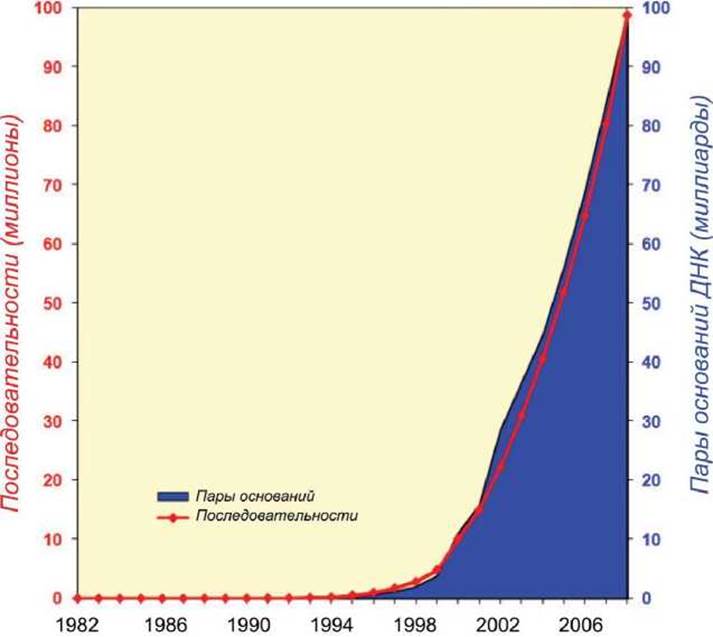
Рисунок 2 - Динамика заполнения базы данных GenBank генетических последовательностей http://www.ncbi.nlm.nih.gov/genbank/genbankstats-2008/
Молекула ДНК состоит из тысяч нуклеотидов, и поэтому определение полной последовательности нуклеотидов целой молекулы хромосомной ДНК представляет собой весьма сложную задачу (см. [6], п. 5). С появлением технологии клонирования генов и полимеразной цепной реакции (ПЦР) учёные получили возможность выделять отдельные фрагменты хромосомной ДНК (см. [7], п. 11). Эти достижения, в свою очередь, проложили путь к развитию быстрых и эффективных методов секвенирования ДНК.
В конце 70-х годов XX века появились два метода секвенирования, основанные, соответственно, на реакциях обрыва цепи и химического расщепления. Эти методы с некоторыми незначительными видоизменениями заложили основу для революции секвенирования 80-х и 90-х годов и последующего рождения биоинформатики.
Благодаря своей чувствительности, специфичности и возможности автоматизации, ПЦР считается передовым методом анализа образцов геномной ДНК и построения генетических карт. Последующие усовершенствования базовой технологии ПЦР дополнительно увеличили мощность и практическую ценность этой методики.
Ещё в начале 80-х годов XX века исследователи вручную (с помощью электронных самописцев) считывали последовательности ДНК с картины полос на гель-плёнке. В 1987 году Стивен Кравец (Stephen А. Krawetz) разработал первое программное обеспечение для устройств автоматического считывания информации с гелиевых плёнок.
С момента получения в 1987 году первой последовательности, секвенированной полуавтоматическим методом, практической реализации ПЦР в 1990 г. и внедрения способа флуоресцентного мечения фрагментов ДНК, производимых методом полимерного копирования по Сангеру (см. [7], и. 11.2), было осуществлено крупномасштабное секвенирование, внесшее неоценимый вклад в развитие биоинформатики. Одновременно значительное развитие получили технологии автоматизированной регистрации результатов секвенирования последовательностей.
В начале 90-х годов Крейг Вентер (John Craig Venter) с сотрудниками изобрёл новый метод определения генов. Вместо того чтобы секвенировать хромосомную ДНК с предельным разрешением в один нуклеотид, группа Вентера выделила молекулы мРНК, копировала их в молекулы кДНК и затем секвенировала некоторую часть молекулы кДНК, в результате чего были созданы ярлыки экспрессируемых последовательностей (expressed sequence tags, EST, термин, впервые предложенный Энтони Керлавейдж (Anthony Kerlavage).
Эти EST-последовательности могли быть использованы в качестве указателей (идентификаторов, "отпечатков пальцев") для выделения целого гена. Кроме того, подход с применением ярлыков EST повлек за собой организацию огромных баз данных нуклеотидных последовательностей и, как полагают, развитие метода EST показало осуществимость
проектов высокопроизводительного обнаружения новых генов и явилось ключевым толчком для развития прикладной геномики.
В 80-х годах XX века начался ряд проектов по созданию подробных генетических и физических карт генома человека (рисунок 3). Цель этих проектов состояла в расшифровке полной последовательности нуклеотидов генома человека и в определении локусов (фиксированных положений, локализации на хромосоме) предполагаемых 30 000 генов. Работа столь большого размаха стимулировала развитие новых вычислительных методов анализа генетических карт и данных секвенирования последовательностей ДНК, а также потребовала разработки новых методов и лабораторного оборудования для расшифровки и анализа ДНК.
Для максимально быстрого ознакомления широкого круга исследователей с результатами расшифровки потребовалось разработать усовершенствованные средства распространения полученной информации.
Международную научно-исследовательскую программу, явившуюся результатом этой глобальной инициативы, назвали проектом "Геном человека" (Human Genome Project, HGP). Более подробную информацию об этом и других проектах расшифровки геномов можно получить по адресам:
✵ http://genomics.energy.gov/;
✵ http://oml.gov/sci/techresources/Human_Genome/publicat/tko/index.html;
✵ http://www.geneontology.org/GO.refgenome.shtml;
✵ http://www.genome.gov/.
В 2007 г. начат проект "1000 геномов" (The 1000 Genome Project) http://www.1000genomes.org - расшифровка полных геномов 1000 человек, каждый содержащий 6 Гига-пар оснований (6 Gbp), а всего 6 Терапар оснований (6 ТЬр) [56]. К марту 2012 г. полное описание расшифрованных генов составило более 250 000 файлов объёмом более 260 Терабайт. Для этого проекта был создан Центр координации данных (DCC, Dato Coordination Center) и были разработаны технологии секвенирования нового поколения (Next-generation sequencing (NGS) technologies) [57], которые снизили стоимость секвенирования одного генома до US$5000.
Рисунок 3 - Веб-страницы геномных проектов: а - Геномной программы Департамента Энергии США; б - То Know Ourselves; в - Проект аннотации геномов; г - Национальный институт исследования генома человека