Основы биоинформатики - Огурцов А.Н. 2013
Информационные принципы в биотехнологии
Анализ и предсказание белков
Предсказание вторичной структуры белка
Одной из важнейших задач анализа последовательностей является точное предсказание очагов формирования а-спиралей, ß-нитей и других элементов вторичной структуры в аминокислотной цепи белка.
Предсказание структуры по аминокислотной последовательности начинают с анализа базы данных известных структур. Поиск в этих базах данных проводят с целью выявления всех возможных связей между последовательностью и структурой белка.
Качество предсказания вторичной структуры зависит от точности распознавания типа элементов вторичной структуры в известных свёртках, а также от точности определения местоположения и протяжённости этих элементов. К основным типам вторичных структур, которые исследуются на предмет изменчивости последовательности, относятся а-спирали, ß-структуры и супервторичные структуры (см. [9], п. 4.2).
В настоящее время наиболее широкое применение нашли следующие методы предсказания вторичной структуры белков:
1) методы Чоу-Фасмена (P.Y. Chou, G.D. Fasman (1974)) и Гарньера- Осгуторпа-Робсона (GOR) (J. Garnier, D.J. Osguthorpe, В. Robson (1978));
2) методы моделирования нейронных сетей;
3) методы поиска "ближайшего соседа".
Метод Чоу-Фасмена, предложенный Питером Чоу (Peter Y. Chou) и Джеральдом Фасменом (Gerald D. Fasman), основан на допущении о том, что каждая аминокислота индивидуально влияет на вторичную структуру в пределах некоторого окна последовательности. Он опирается на анализ частот появления каждой из 20 аминокислот в а-спиралях, и ß-нитях и ß-изгибах. Кроме того, построенный на нём алгоритм использует специальный набор правил предсказания вторичной структуры. Вначале алгоритм просматривает последовательность и пытается найти короткую подпоследовательность аминокислот, показывающую высокую склонность к образованию зародыша структуры определённого типа.
Предсказание а-спиралей считают достаточно правдоподобным, если четыре из шести аминокислот имеют высокую вероятность (>1,0) пребывания в а-спирали. Что касается предсказания склонности к образованию зародыша ß-нити, то его принимают за верное, если на каждые пять аминокислот в последовательности приходится три с вероятностью нахождения в ß-нити > 1,0. Затем алгоритм продолжает области зародышеобразования в обе стороны - до тех пор, пока значения вероятности для группы из четырёх аминокислот не падают ниже 1.
Если для некоторого отрезка последовательности могут быть предсказаны как а-спиральные, так и ß-структурные области, то принимается предсказание с более высокой вероятностью.
Предсказание ß-изгибов (рисунок 81), обеспечивающих поворот пептидной цепи на угол около 180 градусов на протяжении отрезка, содержащего 4 аминокислотных остатка (остатки 1 и 4 соединены водородной связью), основано на несколько ином принципе.
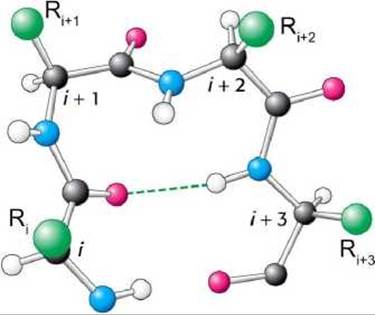
Рисунок 81 - Схема ß-изгиба
Алгоритм моделирует изгибы в виде тетрапептидов и рассчитывает две вероятности.
Во-первых, как и в случае предсказаний а-спиралей и ß-нитей, алгоритм вычисляет среднее от вероятностей пребывания в изгибе каждой из четырёх аминокислот.
Во-вторых, определяются вероятности появления в изгибе аминокислотных комбинаций, начинающихся с очередной позиции тетрапептида.
Затем алгоритм перемножает эти четыре вероятности, вычисленные для группы из четырёх аминокислот в смоделированной последовательности, и находит вероятность того, что данный тетрапептид является изгибом.
Предсказание изгиба считается верным, если значение первой вероятности превышает вероятность появления в данной области а-спирали или ß-нити и если значение второй вероятности больше 7,5∙10-5.
Метод GOR основывается на допущении о том, что аминокислоты, примыкающие к центральному аминокислотному остатку, тоже влияют на вторичную структуру, к которой центральный остаток, вероятно, склонен. При составлении предсказаний этот метод опирается на принципы теории информации. Алгоритм GOR просматривает известные вторичные структуры и определяет частоту появления определённых аминокислот в каждом типе структуры. Кроме того, определяется также частота появления аминокислот (20-ти видов) в восьми соседних позициях (отстоящих от N- и С-концов центральной аминокислоты), и, таким образом, общее количество исследуемых позиций равно 17, включая центральную.
Предсказание с помощью нейронных сетей. Нейронные сети - это класс общих вычислительных структур, которые моделируют анатомию и физиологию биологических нервных систем. Они с успехом применяются к широкому спектру задач распознавания образов, классификации и задачам принятия решений.
В вычислительной схеме одиночный "нейрон" является вершиной графа с одним или несколькими входящими ребрами (входами) и одним исходящим ребром (рисунок 82(a)).
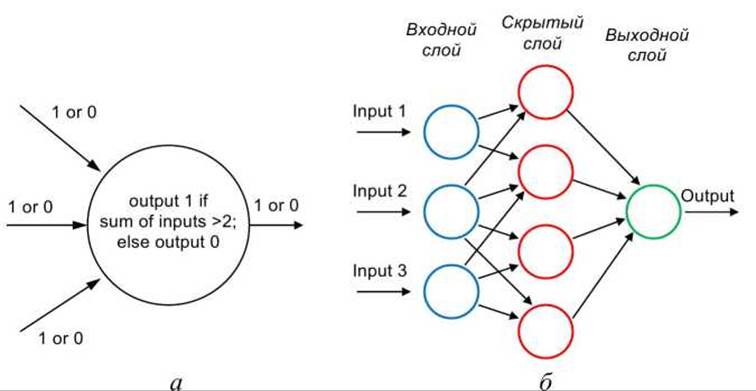
Рисунок 82 - Нейронные сети: а - вершина графа; б - схема простой нейросети
Используя физиологическую метафору, можно сказать, что нейрон испускает сигнал, если на выходе у него 1, и не испускает сигнал, если на выходе 0. Модельные нейроны могут различаться по количеству входов и выходов и по формуле, которая вычисляет выход (рисунок 82(6)).
Чтобы сформировать сеть, необходимо создать несколько нейронов и соединить выходы одних нейронов с входами других нейронов. Некоторые вершины содержат входы для всей сети (область ввода или входной слой, input layer), а некоторые имеют выходы наружу (область выхода или выходной слой, output layer). Кроме того есть нейроны, которые не связаны напрямую с внешним миром (скрытая область или скрытый слой, hidden layer) (рисунок 82(6)).
Неограниченная сложность возможна при создании и соединении нейронов и при определении строгости связей, то есть вместо того, чтобы просто суммировать входные сигналы i1 + i2 + i3, можно использовать взвешенные суммы входов, например 9i1 + 5i2 + i3, что сделает сеть более чувствительной к входу номер 1 и менее чувствительной к входу номер 3. Биологически это соответствует изменению силы синапсов. (Сила синапса - это величина изменения трансмембранного потенциала в результате активации постсинаптических рецепторов нейромедиаторами).
Свойство нейронной сети, которое определяет её мощь, заключается в том, что веса могут рассматриваться как переменные и могут вычисляться в процессе обучения для частных случаев. Чтобы обучить нейронную сеть, её применяют к разным примерам и сравнивают выход с правильным решением. Если ответ не совпадает, то проводят уточнение параметров. В процессе обучения топология сети остается неизменной, при этом, если вес какой-либо связи оказывается равным 0, то это эквивалентно разрыву соответствующей связи.
Тип нейронной сети, которая может быть применена к распознаванию вторичной структуры белка, показан на рисунке 83.
Входная область (IL, input layer) сканирует последовательность окном в 15 остатков, то есть анализируется фрагмент последовательности размером 15 элементов. Предсказание относится к центральному остатку (наверху, отмечен стрелкой). Затем окно сдвигается на одну позицию вправо по последовательности и делается следующее предсказание, Каждой из 15 позиций (аминокислот) в окне соответствует 20 нейронов, один из которых активен (чёрный кружок).
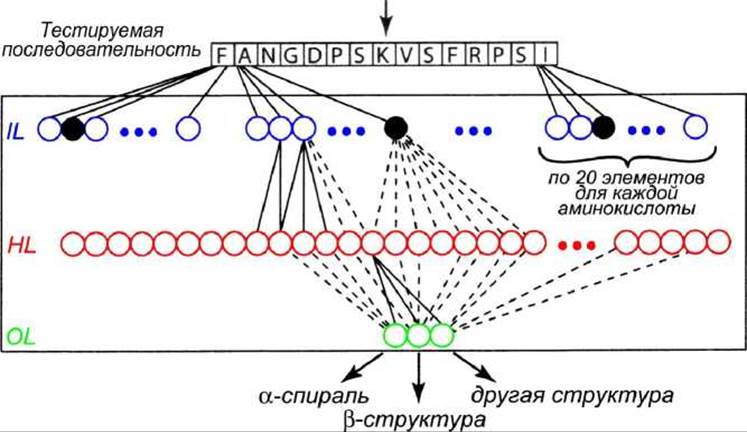
Рисунок 83 - Нейронная сеть для предсказания вторичной структуры белка
Скрытая область (HL, hidden layer) содержит 15x20=300 нейронов, соединённых с вводом и выводом. Каждый нейрон в скрытой области соединён с каждым нейроном областей ввода и вывода (на рисунке показаны не все связи).
Область вывода (OL, output layer) состоит только из трёх нейронов, которые просто фиксируют предсказание - а-спираль, ß-структура, или ни то ни другое.
Важной информацией, которая может быть использована при предсказании вторичной структуры, является эволюционная информация. Множественное выравнивание содержит в себе гораздо больше информации, чем одна последовательность. Сохранение вторичной структуры в родственных белках означает наличие связи последовательность-структура, и это позволяет назначать им более высокие профильные веса и делать более строгие предсказания. Большинство методов предсказания вторичных структур, основанных на нейронных сетях, имеют на входном слое не только информацию о степени консервативности позиции, но и профильные веса.
Кроме того, использование двух тандемных (следующих друг за другом) нейронных сетей позволяет учитывать корреляцию конформаций соседних остатков. Предсказания состояний нескольких последовательно идущих остатков с помощью сети, аналогичной показанной на рисунке 83, комбинируется с помощью ещё одной сети, которая формирует окончательный результат.
В подходе нейронных сетей компьютерные программы обучаются распознавать регулярные комбинации аминокислот, находящиеся в известных вторичных структурах, и отличать эти комбинации от других аминокислотных групп, не пребывающих в этих структурах. Такие модели нейронной сети большую часть информации из последовательностей извлекают путём алгоритмической интерпретации.
Из программ моделирования нейронных сетей можно назвать
- PHD (http://www.predictprotein.org/);
- NNPREDICT (http://www.cmpharm.ucsf.edu/~nomi/nnpredict.html).
Предсказание методом поиска ближайшего соседа. Так же как и методы нейронных сетей, методы поиска ближайшего соседа основаны на принципе обучающейся машины. Они предсказывают предпочтение аминокислоты из последовательности запроса к определённой конформации вторичной структуры. Для этого алгоритм сравнивает последовательность запроса с подобными ей последовательностями, структура которых известна.
Алгоритм передвигает окно переменной длины по набору из 100— 400 обучающих последовательностей с известной структурой и составляет большой список коротких фрагментов последовательностей - кандидатов на тестирование.
Затем отмечается минимальное взаимное подобие последовательностей, а также вторичная структура, соответствующая центральной аминокислоте в каждом из окон. После этого алгоритм выбирает из последовательности запроса окно того же самого размера, сравнивает его с каждым из вышеупомянутых фрагментов-кандидатов и определяет 50 фрагментов, дающих наилучшие совпадения. Наконец, по частотам появления известной вторичной структуры средней аминокислоты в каждом из этих 50 фрагментов строится предсказание вторичной структуры средней аминокислоты в окне последовательности запроса.
Склонность к формированию вторичной структуры. В своё время было предпринято множество попыток предсказать вторичную структуру прямо по последовательности аминокислот. Наблюдения над растворами образцовых полипептидов показали, что аминокислоты обладают сильной изменчивостью в склонности к образованию регулярных конформаций.
Самые первые попытки предсказания вторичной структуры базировались на параметризации физических моделей. Так, в ходе физикохимических исследований образцовых полипептидов было установлено, что склонность аминокислоты к продолжению спиралей может отличаться от её же склонности к зарождению таковых.
Как сказано выше, Чоу и Фасмен предложили подход, основанный на статистической модели. Согласно этому подходу частоту появления определённой аминокислоты в некоторой конформации сравнивают со средним значением частот появления в этой же конформации всех известных аминокислот (из алфавита). Полученное отношение выражает склонность этой аминокислоты к появлению в данной конформации. По этим величинам аминокислоты были классифицированы на различные классы, и на их же основании были сформулированы правила предсказания вторичной структуры.
Методы Чоу-Фасмена и GOR опираются на концепцию склонности аминокислот к формированию определённых вторичных структур. Как оказалось, аминокислоты оказывают предпочтение некоторым состояниям вторичной структуры, что отражено в таблице 20.
Значение вероятности 1,0 указывает на то, что склонность данной аминокислоты к соответствующей вторичной структуре равна усредненной склонности всех известных аминокислот; превышающие единицу значения указывают на склонность выше средней; значения меньше единицы указывают на склонность ниже средней.
Таблица 20 - Склонность аминокислот к формированию а-спиралей и ß-структур
|
Аминокислота |
а-спираль |
ß-струкіура |
|
Glu (Е, глутаминовая кислота) |
1,59 |
0,52 |
|
Ala (А, аланин) |
1,41 |
0,72 |
|
Leu (L, лейцин) |
1,34 |
1,22 |
|
Met (V, метионин) |
1,30 |
1,14 |
|
Gln (Q, глутамин) |
1,27 |
0,98 |
|
Lys (К, лизин) |
1,23 |
0,69 |
|
Arg (R, аргинин) |
1,21 |
0,84 |
|
His (Н, гистидин) |
1,05 |
0,80 |
|
Val (V, валин) |
0,90 |
1,87 |
|
Ile (I, изолейцин) |
1,09 |
1,67 |
|
Tyr (Y, тирозин) |
0,74 |
1,45 |
|
Cys (С, цистеин) |
0,66 |
1,40 |
|
Trp (W, триптофан) |
1,02 |
1,35 |
|
Phe (F, фенилаланин) |
1,16 |
1,33 |
|
Thr (Т, треонин) |
0,76 |
1Д7 |
|
Gly (G, глицин) |
0,43 |
0,58 |
|
Asn (N, аспарагин) |
0,76 |
0,48 |
|
Pro (Р, пролин) |
0,34 |
0,31 |
|
Ser (S, серин) |
0,57 |
0,96 |
|
Asp (D, аспарагиновая кислота) |
0,99 |
0,39 |
Значения рассчитаны путём деления частоты встречаемости данного остатка в соответствующей вторичной структуре на среднее значение частот появления в этой вторичной структуре всех известных остатков. Например, глутаминовая кислота отдает явное предпочтение спиральной вторичной структуре (1,59 против 0,52), а глицин имеет склонность ниже среднего (0,43 и 0,58) к обоим типам регулярной вторичной структуры, что говорит о его тяготении к петлям.
Однако точность этих ранних методов, основанных на местном составе аминокислот отдельных последовательностей, была довольно низка и позволяла предсказать правильное состояние вторичной структуры не более чем 60 % остатков.
Собственное стремление аминокислот к формированию ß-изгибов. Частота появления пар аминокислот в ß-изгибах была вычислена по данным анализа результатов исследования кристаллической структуры белка. Были отмечены следующие частоты: Pro-Asn (63%), Pro-Phe (50%), Pro-Gly (38%), Pro-Ser (31%) и Pro-Val (8%). Однако статистический анализ, основанный на ином критерии оценки склонности к ß-изгибам, показал существенное различие в порядке предпочтений. Так, при исследовании набора белковых структур из базы данных был определен следующий порядок предпочтений: Pro-Gly > Pro-Asn > Pro-Ser > Pro-Val > Pro-Phe.
Склонность к формированию ß-изгибов оценивалась путём измерения стандартной свободной энергии Гиббса циклизации пептида в образцовых тетрапептидах типа Cys-Pro-X-Pro. Был отмечен следующий порядок предпочтений: Pro-Asn > Pro-Gly > Pro-Ser > Pro-Phe > Pro-Val.
ЯМР-измерения температурной зависимости химических сдвигов в образцовых пептидах типа Tyr-Pro-X-Asp-Val позволили определить семейства ß-изгибов. Согласно данным ЯМР-спектроскопии, семейства ß-изгибов стоят в порядке Pro-Gly > Pro-Asn > Pro-Phe > Pro-Ser > Pro-Val.
Совместный анализ данных термодинамики, ЯMP и (статистических) данных о кристаллической структуре показывает, что порядок предпочтений выглядит как: Pro-Gly, Pro-Asn > Pro-Ser > Pro-Val.
Хотя относительное положение пары Pro-Phe кажется очень изменчивым в этом ряду, для других пептидов наблюдается чёткая корреляция между статистическими предпочтениями, рассчитанными по белковым структурам из базы данных и предпочтениями, основанными на данных термодинамического анализа и ЯМР-спектроскопии образцовых соединений.
Библиотеки ротамеров. Ротамер (rotamer) определяется как один из ряда конформеров, являющийся результатом ограниченного вращения вокруг одной единственной химической связи.
Конформер (conformer) или поворотный изомер - это особое свёрнутое состояние или структура белка, обладающее низкой энергией, то есть термодинамически устойчивое при физиологических условиях.
Исследование конформационных предпочтений боковых цепей показало, что несколько конформационных вариантов боковых цепей имеют намного более высокую вероятность существования, чем другие. Эти результаты побудили учёных к проведению целого ряда исследований, призванных определить функцию вероятности того, что данная боковая цепь, присущая данной аминокислоте, появится в соответствующей конформации, а также установить зависимость конформации боковой цепи от конформации основной цепи.
Благодаря значительно возросшему объёму баз данных, было создано множество библиотек ротамеров. Библиотеки ротамеров могут быть использованы в молекулярном моделировании: алгоритм извлекает из них наиболее вероятные конформации боковых цепей и добавляет их к основной цепи макромолекулы.