Основы биоинформатики - Огурцов А.Н. 2013
Информационные принципы в биотехнологии
Биоинформатика в фармации
Фармакоинформатика
Термин фармакоинформатика часто употребляют для описания дисциплины, объединяющей в себе биологию, химию, математику и информационные технологии с целью обработки и анализа данных в фармацевтической промышленности.
Применение высокопроизводительных отборочных испытаний в открытии лекарственных препаратов зависит от наличия разнообразных химических библиотек (например, создаваемых методами комбинаторной химии), так как они значительно увеличивают возможности отыскания молекул, взаимодействующих с определённой белковой мишенью.
Количественно определить химическое разнообразие очень сложно. Так, были предприняты попытки решения этой проблемы с помощью концепции "химического пространства". По сути, химическое пространство заключает в себе химическое соединение со всеми возможными химическими свойствами, сосредоточенными во всех потенциально активных участках молекул. Таким образом, библиотека с высоким показателем разнообразия будет обладать широким охватом химического пространства, не содержащего промежутки и группы подобных молекул.
Количественно разнообразие библиотек обычно определяют с помощью величин, основанных на сравнении свойств различных молекул, описываемых такими параметрами, как расположение и заряд атомов, а также способность к формированию различных типов химической связи.
Для сравнения двух молекул можно использовать коэффициент Танимото КТ, отражающий степень подобия фрагментов этих молекул. Коэффициент Танимото вычисляют по формуле
![]()
где а - число параметров фрагментов в соединении А; b - число параметров фрагментов в соединении В, с - число общих (подобных) параметров фрагментов этих соединений. Следовательно, для идентичных молекул КT = 1, тогда как для молекул без общих параметров КT = 0. В химической библиотеке с идеальным показателем разнообразия большая часть попарных сравнений дала бы коэффициент Танимото, близкий к нулю.
В том случае, если о специфичности связывания белка-мишени почти ничего не известно, продуктивное открытие лидов могут обеспечить максимально объёмные химические библиотеки с высоким показателем разнообразия. Если же о последовательности или о структуре мишени удалось собрать информацию строго определённого вида, то можно из общих библиотек выбрать частные библиотеки, охватывающие какую-либо одну область химического пространства.
Например, если известна последовательность некоторого белка-мишени, то поиск гомологии в базе данных часто будет давать родственный белок с ранее установленной структурой и уже описанными взаимодействиями с маленькими молекулами.
В таких случаях возможно спроектировать химическую библиотеку, содержащую один молекулярный каркас, который сохраняет относительное расположение участков, присутствующих в известном лиганде, но который может быть видоизменен путём прикрепления к нему разнообразных функциональных групп. Вполне возможно, что ранее уже было показано, что некоторые из этих групп необходимы для связывания лекарственных препаратов. Такие участки называют фармакофорами.
Термин фармакофор (pharmacophore) был введен Паулем Эрлихом (Paul Ehrlich) в 1909 году. Эрлих определил фармакофор, как молекулярный остов, который несёт (phore) существенные признаки, ответственные за биологическую активность лекарства (pharmaco).
В 1977 году это определение было модифицировано Питером Гундом (Peter Gund): фармакофор - это набор структурных признаков в молекуле, которые распознаются биологическими рецепторами и являются ответственными за биологическую активность молекулы.
Современное определение ИЮПАК (IUРАС, The International Union of Fure and Applied Chemistry) таково: фармакофор - это набор пространственных и электронных признаков, необходимых для обеспечения оптимальных супрамолекулярных взаимодействий со специфической биологической мишенью, которые могут вызывать (или блокировать) её биологический ответ.
Под фармакофорными признаками обычно понимаются фармакофорные центры и интервалы расстояний между ними, необходимые для проявления данного типа биологической активности.
Типичными фармакофорными центрами при этом являются: гидрофобные области, ароматические кольца, доноры и акцепторы водородной связи, анионные и катионные центры.
Для более детального описания фармакофора часто используют гидрофобные и исключённые объёмы, а также допустимые интервалы угловой ориентации векторов водородных связей и плоскостей ароматических колец.
При фармакофорном поиске проводится поиск соответствия между фармакофорной моделью и характеристиками молекул из базы данных, находящихся в допустимых конформациях.
Пример фармакофора приведён на рисунке 85.
Функциональные группы фармакофора изображены на рисунке 85 в виде сфер разного радиуса. Компьютерные программы используют разные цвета для отображения сфер, соответствующих гидрофобным и гидрофильным областям, или же положительно и отрицательно заряженным функциональным группам. Узконаправленные водородные связи изображены конусами. Соответствующая молекула лида вписана в фармакофорную модель.
Фармакофорный скрининг позволяет значительно сократить время поиска необходимых лидов. Прежде чем проводить отсеивающие эксперименты в лабораторных условиях, имеет смысл попытаться собрать как можно больше информации о потенциальных взаимодействиях препарата с мишенью.
Одним из путей получения таких данных является автоматический отборочный поиск в химических базах данных (на соответствие молекулярной мишени с известной структурой).
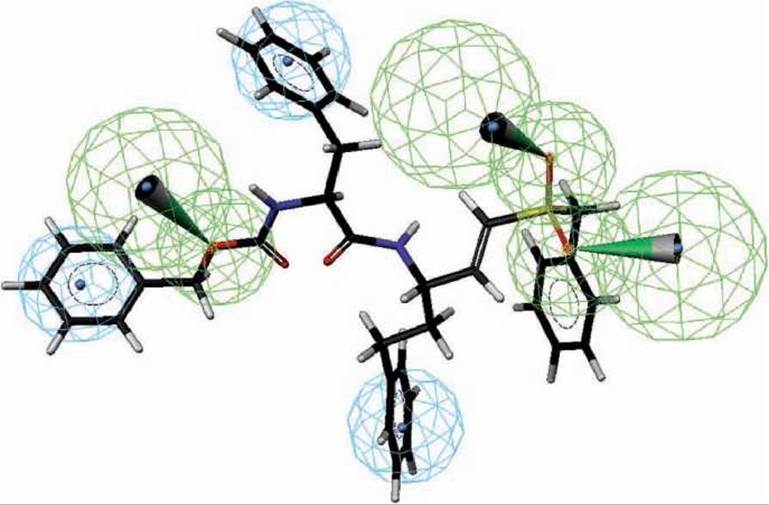
Рисунок 85 - Модель фармакофора с молекулой лида
В других случаях структуру соединения можно попытаться определить по подобию с установленной структурой близкой гомологии или предсказать её с помощью алгоритма трединга (протягивания).
Если структура белка-мишени известна, то применяют основанные на критерии адекватности алгоритмы распознавания потенциальных взаимодействующих лигандов, так называемые "рациональные", прямые методы компьютерного конструирования лекарств на основании структуры белка-мишени (structure-based drug design).
Сначала устанавливают место связывания (рецептор) низкомолекулярного соединения (лекарства) с белком-мишенью.
Затем используют докинг (docking, стыковка) - производят молекулярно-графический анализ комплекса лиганд—рецептор - возможность размещения потенциального лиганда в полости активного центра макромолекулы и оценку энергии связывания и сродства для такого комплекса. Конструирование новых лигандов можно также осуществить посредством модификации структуры найденных соединений.
К настоящему времени разработано большое число алгоритмов докинга, которые пытаются подобрать маленькие молекулы к участкам связывания, анализируя информацию о пространственных ограничениях и энергии связей. Наиболее популярные программы молекулярного докинга доступны через Интернет:
|
AutoDock |
http://autodock. scripps.edu/ |
|
FlexX |
http://www.biosolveit.de/FlexX/index.html?ct=l |
|
Hex |
http://hex.loria.fr/ |
|
Dick Vision |
http://dockvision.com/ |
|
eHiTS |
http://www.simbiosys.ca/ehits/index.html |
|
FRED |
http://www.eyesopen.com/fred |
|
GOLD |
http://www.ccdc.cam.ac.uk/products/life_sciences/gold/ |
|
LIGPLOT |
http://www.ebi.ac.uk/thornton-srv/software/LIGPLOT/ |
|
Pocket-Finder |
http://www.modelling.leeds.ac.uk/pocketfmder/ |
|
Q-SiteFinder |
http://www.modelling.leeds.ac.uk/qsitefmder/ |
|
SITUS |
http://situs.biomachina.org/index.html |
|
Molecular Doc king Web |
http://mgl.scripps.edu/people/gmm/ |
На рисунке 86 приведен пример визуализации результатов докинга молекулы цитостатического лекарственного препарата дактиномицин (актиномицин D, из группы противоопухолевых антибиотиков, подгруппы актиномицинов), который используется в качестве паллиативной терапии некоторых типов рака (облегчающих течение болезни у пациента).
Программа докинга рассматривает каждый потенциальный лиганд как каркас, с присоединёнными функциональными группами. Сначала алгоритм предсказывает возможные варианты стыковки путём анализа расположения ван-дер-вальсовых сфер (только тех, которые находятся на каркасе), после чего проверяет на стерическую совместимость отдельные функциональные группы, используя разнообразные сочетания вращений связей. Наконец, алгоритм производит стыковку и вычисляет общий счёт комплекса.
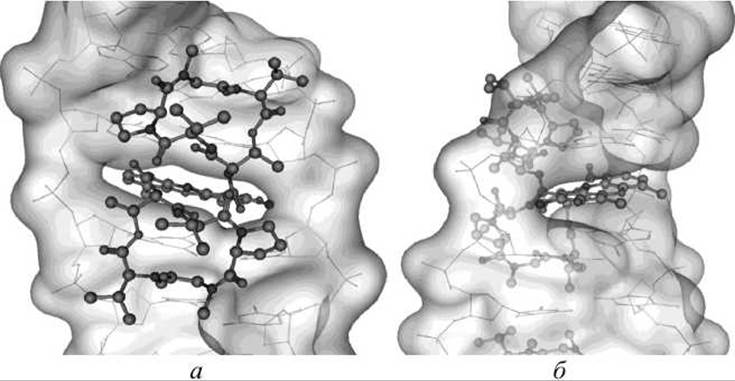
Рисунок 86 - Докинг молекулы дактиномицина интеркалировавшего между парами оснований молекулы ДНК: а - вид спереди; б - вид сбоку
В химических базах данных можно проводит поиск не только на соответствие участку связывания (поиск взаимодействий комплементарных молекул) но также и на соответствие некоторому лиганду. Известно несколько алгоритмов сравнения двумерных или трёхмерных структур и построения профилей подобных молекул.
Установление трёхмерной структуры мишени (рентгеноструктурный анализ, ЯМР-интраскопия) является необходимым условием разработки лида, который должен или связываться с ней, или воздействовать на неё.
Лиды выбирают из существующей библиотеки химических соединений путём комбинаторной стыковки структур. Варианты лидов из библиотеки поочередно состыковывают с активным участком молекулярной мишени (путём перебора вариантов комплементарного связывания). Эта предварительная "подгонка" in silico сокращает число соединений, которые необходимо синтезировать и испытывать in vitro, так как базы данных содержат необходимые (для имитационного моделирования) описания химических свойств и методов синтеза соединений.
Другой метод поиска лидов конструирует молекулы из библиотеки функциональных групп на основе анализа сайта связывания на поверхности мишени. Специальный алгоритм имитационного моделирования тщательно анализирует активный участок молекулярной мишени и выстраивает молекулу лида из отдельных фрагментов.
Поверхность молекулярной мишени, которая должна взаимодействовать с лидом, может соседствовать с различными по химическим свойствам областями белковой молекулы, например, с зонами гидрофобности, с зонами образования водородных связей или с каталитическим активным центром. В эти области последовательно помещают фрагменты гипотетического соединения. Оптимизация ориентации фрагментов помогает подобрать финальную пространственную структуру лида.
Иногда целая молекула сразу вписывается в рецепторный или активный участок и программа докинга перебирает все возможные способы подогнать лиганд к рецепторному участку. Участок связывания в молекуле рецептора или фермента содержит области образования водородных связей, а также гидрофобные области.
Первоначально программа помещает и ориентирует молекулу-прототип в активном участке таким образом, чтобы образовалось максимально возможное количество связей.
Затем последовательно добавляет и подгоняет дополнительные функциональные группы до тех пор, пока не будут сформированы все необходимые связи лиганда с мишенью.
Программа моделирует варианты расположения элементов активного участка мишени и затем ищет в базе данных химические структуры, которые удовлетворяли бы такой имитации.
Если нет данных о пространственной структуре белка, тогда при компьютерном конструировании применяются разнообразные методы сравнительного моделирования. Функционально важные участки в молекуле белка удаётся выявить путём сравнительного анализа аминокислотных последовательностей гомологичных белков.
Для поиска в базах данных секвенированых последовательностей и в структурных базах данных используется программа BLAST. При построении трёхмерной модели белка с заданной аминокислотной последовательностью эта полипептидная цепочка вначале "вписывается" в координаты, соответствующие аминокислотным остаткам гомологичного белка с расшифрованной пространственной структурой, а затем осуществляется минимизация внутренней энергии, чтобы убрать возможные напряжения в структуре.
После этого методами молекулярной динамики моделируется движение отдельных частей молекулы, чтобы уточнить расположение гибких участков. Качество модели оценивается с помощью программы, сравнивающей расположение аминокислотных остатков с известной статистикой для белков, пространственная структура которых была расшифрована экспериментально.
При анализе взаимодействия лиганд-рецептор следует принимать во внимание структурную подвижность молекулы лиганда. Чтобы произошло связывание лиганда, его молекула должна находиться в такой конформации, которая комплементарна структуре белка. Статическая модель взаимодействия лиганд-рецептор не учитывает конформационной подвижности. Конформационное пространство (набор конформационных вариантов) лиганда оценивается с помощью моделирования молекулярной динамики и минимизации энергии. Производится докинг возможных конформации лиганда в разных положениях. Наилучшие положения используются для молекулярно-динамического моделирования комплекса лиганд-белок.
Результаты моделирования показывают, при каких состояниях рецептор наиболее часто связывается с конформационными вариантами лиганда. Моделирование структуры рецептора в присутствии и в отсутствие лиганда обеспечивает сведения о том, как изменяется конформация белка при его активации, обусловленной взаимодействием с лигандом.
Уже известны десятки лекарственных средств, сконструированных с применением технологий биоинформатики, которые успешно прошли клинические испытания и введены в медицинскую практику.
Описанный метод разработки лекарственных препаратов, при котором опытные соединения оптимизируют путём добавления к молекулярному каркасу различных функциональных групп и проверки каждого производного соединения на его биологическую активность, должен ещё включать в себя процедуру анализа структура-активность. Иначе, если на моделируемой молекуле есть множество открытых позиций, которые можно заместить функциональными группами, то общее количество молекул, которые должны быть впоследствии проверены при всестороннем отборочном анализе, является слишком большим.
Синтез и тестирование всех этих молекул потребовали бы значительных временных затрат и производственных усилий, хотя очевидно, что большинство этих молекул-кандидатов не обладает никакой полезной функцией.
Методы анализа количественных взаимосвязей структура-активность (Quantitative Structure-Activity Relationship, QSAR) с двумерным (2D) или трёхмерным (3D) представлением структуры лигандов, сравнительным анализом молекулярных полей (Comparative Molecular Field Analysis, CoMFA), сравнительным анализом молекулярного подобия (Comparative Molecular Similarity Indices Analysis, CoMSIA), и голографическая количественная связь структура-активность (Hologram Quantitative Structure Activity Relationship, HQSAR) обеспечивают пространственное картирование места связывания лиганда, построение модели фармакофора и проведение скрининга потенциальных лигандов в химических базах данных.
Оценка по QSAR позволяет отобрать только те молекулы, которые с наиболее высокой вероятностью будут иметь полезную активность, и таким образом уменьшить число молекул-кандидатов для последующего химического синтеза.
QSAR представляет собой выраженное в математической форме отношение, которое описывает взаимосвязь структуры молекулы с её биологической активностью.
Молекулы рассматриваются как совокупности молекулярных свойств (параметров), организованных в виде таблицы. Программа QSAR просматривает эти данные и пытается находить совместимые отношения между отдельными параметрами и биологическими функциями и таким образом определить набор правил, которые могут быть использованы для назначения счёта новым молекулам при оценке их потенциальной активности. QSAR обычно выражают в виде линейного уравнения
![]()
где А - биоактивность; Pi - параметры (молекулярные свойства), установленные для каждой из N молекул набора; С, - коэффициенты, рассчитываемые путём подгонки параметров молекул к их биологическим функциям.
Как только набор лидов определён, молекулы должны быть оптимизированы по эффективности действия, избирательности и фармакокинетическим свойствам. О высокой биологической усвояемости (всасывании в желудочно-кишечном тракте) говорит наличие следующих четырёх качеств:
1) число доноров водородной связи < 5;
2) число акцепторов водорода < 10;
3) относительный молекулярный вес < 500;
4) липофильность < 5.
Лекарства, нацеленные на центральную нервную систему, должны, кроме того, обладать достаточной проницаемостью гемато-энцефалического барьера (blood-brain barrier, ВВВ).