Молекулярная биология клетки - Том 1 - Албертс Б., Брей Д., Льюис Дж., Рэфф М., Робертс К., Уотсон Дж. 1994
Молекулярная организация клеток
Основные генетические механизмы
Клонирование ДНК и генная инженерия
Открытия, о которых шла речь в этой главе, были порождены стремлением ученых постичь жизнь клеток и основные механизмы наследственности. Однако в последние годы эти фундаментальные знания получили практическое применение. Методы клонирования ДНК и генная инженерия дают возможность выделять те или иные гены в достаточном количестве, «перекраивать» их по своему усмотрению и затем вновь вводить в какие-нибудь клетки и организмы. Эти методы составляют лишь часть того общего набора методов, который известен как технология рекомбинантных ДНК (о нем мы уже говорили в гл. 4). Появление технологии рекомбинантных ДНК вызвало подлинную революцию в науке о живых клетках. Кроме того, оно открыло перед медициной и промышленностью новые пути к получению в достаточном количестве тех белков, которые раньше либо были недоступны вообще, либо могли быть получены лишь в очень малых количествах.
5-48
5-49
5-50
5.6.1. Рестрицирующие нуклеазы облегчают клонирование генов [55]
В 1960-е годы возможность выделить в изолированном виде отдельный ген казалась бесконечно далекой. Ген в клетке в отличие от белка не представляет собой некой дискретной единицы; он есть не что иное, как небольшой участок во много раз большей молекулы ДНК. И хотя механическим воздействием молекулы ДНК в клетке можно разделить на множество сегментов, это разделение происходит по закону случая, и фрагмент, содержащий интересующий нас ген может затеряться среди миллиона других фрагментов. Как получить нужный ген в очищенном виде? Поскольку все молекулы ДНК состоят из одних и тех же четырех видов нуклеотидов, которые входят в их состав примерно в равных пропорциях, их нельзя легко разделить подобно тому, как это делают с белками, на основе различий в их зарядах или по их способности связываться с теми или иными реагентами (см. разд. 4.4.3). Более того, если бы даже какую-нибудь схему очистки и можно было предложить, понадобилось бы очень много ДНК для того, чтобы получить нужный ген в количестве, достаточном для последующих экспериментов. Решение всех этих проблем стало реальным, когда были открыты ферменты, названные рестрицирующими нуклеазами (рестриктазами). Эти ферменты, которые можно выделить в очищенном виде из бактериальных клеток, разрезают двойную спираль ДНК по специфическим последовательностям из 4-8 нуклеотидов, разделяя ее на фрагменты строго определенного размера (их называют рестрикционными фрагментами). Специфичность рестрицирующих нуклеаз в отношении нуклеотидных последовательностей у разных видов бактерий различна, так что не очень трудно подобрать такую рестриктазу, которая вырежет небольшой фрагмент ДНК, содержащий определенный ген. Размер этого рестрикционного фрагмента и послужит затем ориентиром для частичной очистки данного гена (выделения его из смеси).
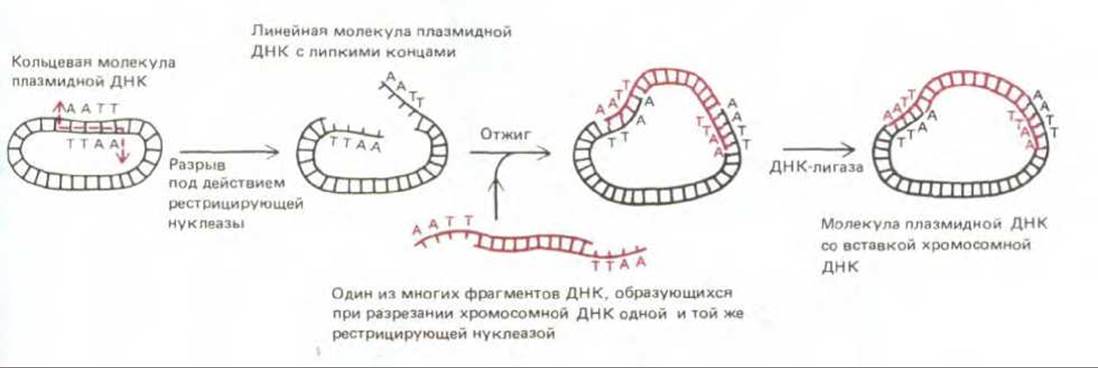
Рис. 5-78. Липкие концы, образующиеся под действием многих рестрицирующих нуклеаз (см. рис. 4-63), обеспечивают возможность соединения двух фрагментов ДНК за счет комплементарных взаимодействий. Фрагменты ДНК, соединившиеся таким путем, связываются затем ковалентно в высокоэффективной реакции, катализируемой ДНК-лигазой. Здесь представлен случай образования рекомбинантной молекулы ДНК, состоящей из плазмиды и встроенной в нее хромосомной ДНК.
Еще одно свойство рестрицирующих нуклеаз делает их удобным инструментом для клонирования генов. Многие из них вызывают ступенчатые разрывы, вследствие чего на обоих концах фрагмента ДНК образуются короткие одноцепочечные «хвосты». Их называют липкими концами, потому что они способны к комплементарному взаимодействию с любым другим концом, образовавшимся под действием того же фермента. Благодаря липким концам, образуемым рестрицирующей нуклеазой, два фрагмента двойной спирали ДНК, происходящие из разных геномов, могут соединиться в одно целое путем спаривания оснований (рис. 5-78). Можно, например, присоединить in vitro фрагмент ДНК, содержащий какой-нибудь ген человека, к хромосоме вируса бактерий и полученную таким путем новую рекомбинантную молекулу ДНК ввести затем в бактериальную клетку. Начав с одной этой рекомбинантной молекулы ДНК, инфицировавшей одну бактериальную клетку, репликационная машина вируса может менее чем за сутки выработать свыше 1012 идентичных молекул вирусной ДНК, а значит, точно в такой же мере размножить и присоединенный к ней фрагмент ДНК человека. Вирус, используемый для подобной цели, называют клонирующим вектором.
5-51
5.6.2. Получение библиотеки ДНК с помощью вирусных или плазмидных векторов [56]
Клонирование того или иного гена начинается с создания библиотеки ДНК в вирусном или плазмидном векторе. Принципы, лежащие в основе клонирования генов, для обоих этих типов векторов одинаковы, хотя в деталях методы могут и различаться. Ради простоты мы в этой главе пренебрежем различиями и проиллюстрируем методы, о которых идет речь, применительно к плазмидам.
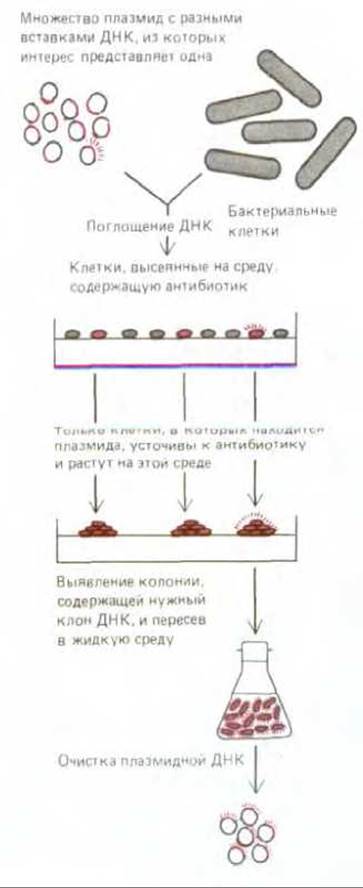
Рис. 5-79. Очистка и амплификация специфической последовательности ДНК путем клонирования ДНК в бактериальных клетках.
Плазмидные векторы, используемые при клонировании генов, представляют собой небольшие кольцевые молекулы двухцепочечной ДНК, происходящие от более крупных плазмид, обычно присутствующих в клетках бактерий, дрожжей и млекопитающих (см. разд. 5.5.11). Как правило, они составляют лишь небольшую фракцию всей ДНК клетки-хозяина, однако благодаря малым размерам их можно легко отделить от молекул хромосомной ДНК, которые, будучи более крупными, при центрифугировании оказываются на дне пробирки. Клонирование начинают с того, что очищенные кольцевые молекулы плазмидной ДНК обрабатывают одной из рестриказ и получают таким путем линейные молекулы ДНК. Затем клеточную ДНК, из которой требуется создать библиотеку, разрезают при помощи той же рестриктазы и полученные рестрикционные фрагменты (включая и те, в которых присутствует клонируемый ген) добавляют к разрезанным плазмидам, а затем подвергают отжигу, для того чтобы могли образоваться рекомбинантные кольцевые молекулы ДНК. Ковалентное сшивание под действием ДНК-лигазы завершает образование этих рекомбинантных молекул, содержащих вставки чужеродной ДНК (см. рис. 5-78).
Следующий шаг к получению библиотеки ДНК состоит в том, что рекомбинантные кольцевые молекулы ДНК вводят в клетки (обычно бактериальные или дрожжевые), сделав их для этой цели временно проницаемыми для ДНК; клетки, как принято говорить, трансфицируют плазмидами. Теперь, когда эти клетки растут и делятся, рекомбинантные плазмиды также реплицируются, в результате чего получается в конце концов огромное количество копий кольцевых молекул, содержащих чужеродную ДНК (рис. 5-79). Многие бактериальные плазмиды несут в себе гены устойчивости к антибиотикам. Это их свойство можно использовать для того, чтобы отделить трансфицированные клетки от тех, которые остались нетрансфицированными: если выращивать бактерии в присутствии антибиотика, то выживут и образуют колонии только те клетки, которые содержат плазмиды. Эти выжившие клетки и заключают в себе библиотеку ДНК. Однако лишь немногие из них несут те рекомбинантные плазмиды, в которых находится подлежащий выделению ген. Надо уметь их идентифицировать, чтобы получить интересующую нас ДНК в очищенном виде и в достаточном количестве. Прежде чем говорить о том, каким способом можно этого достичь, нам следует описать и другой путь получения библиотеки ДНК, также используемый при клонировании.
5.6.3. Два типа библиотек ДНК используются для разных целей [57]
Описанное выше в качестве одного из этапов клонирования разрезание генома на фрагменты с помощью рестрицирующей нуклеазы называют иногда «методом дробовика» (шотган-клонирование). Фрагменты ДНК образуются при этом в огромном количестве - до миллиона, если речь идет о геноме млекопитающих, а это значит, что и число различных колоний трансфицированных клеток также должно достигать миллионов. Каждая такая колония представляет собой клон, т. е. совокупность потомков одной клетки-родоначальницы, и рекомбинантная плазмида в любой клетке клона несет одну и ту же включенную в нее нуклеотидную последовательность геномной ДНК. Плазмиды одной колонии содержат клон геномной ДНК, а вся совокупность плазмид вмещает библиотеку геномной ДНК. Однако, поскольку разрезание геномной ДНК на фрагменты определяется случаем, лишь некоторые образовавшиеся фрагменты содержат полноценные гены; во многие из них попадает только часть какого- нибудь гена, а в большинстве клонов геномной ДНК, полученных из ДНК высших эукариотических клеток.
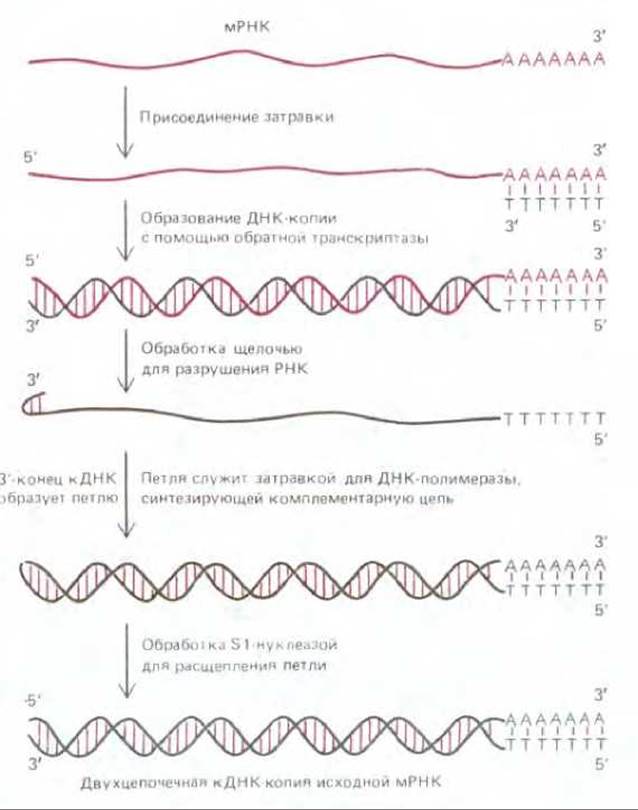
Рис. 5-80. Синтез кДНК. Обратная транскриптаза синтезирует ДНК-копию (кДНК) молекулы мРНК, образуя таким путем гибридную спираль ДНК/РНК. Под действием щелочи РНК-цепь этой гибридной спирали распадается на отдельные нуклеотиды, после чего оставшаяся одноцепочечная кДНК копируется ДНК-полимеразой с образованием двухцепочечной кДНК. Как видно из схемы, для начала синтеза обоим ферментам необходима затравка. Для обратной транскриптазы затравкой служит небольшой олигонуклеотид, в данном случае к длинной последовательности polyA, имеющейся на 3'-конце почти всех мРНК, присоединяется oligo(dT).
Отметим, что у образовавшейся здесь двухцепочечной молекулы кДНК отсутствуют липкие концы; такие молекулы ДНК, с «тупыми» концами, можно клонировать при помощи нескольких процедур, сходных с той, какая представлена на рис. 5-78, хотя и менее эффективно. Можно, например, пришить к концам кДНК синтетические олигонуклеотиды, содержащие участки, в которых рестрицирующий фермент вызывает разрыв, или присоединить ферментативным путем к концам молекулы кДНК одноцепочечные «хвосты», чтобы облегчить включение этой молекулы в клонирующий вектор присутствует лишь некодирующая ДНК, составляющая, как известно, большую часть массы такого генома (см. разд. 9.1.3).
Альтернативный метод начинает процесс клонирования с отбора тех последовательностей ДНК, которые транскрибируются в РНК и, значит, предположительно соответствуют генам. Осуществляется это путем извлечения из клеток мРНК (или очищенной субфракции мРНК) и затем получения комплементарной ДНК-копии (кДНК) каждой наличной молекулы мРНК; эта реакция катализируется обратной транскриптазой- ферментом ретровирусов, синтезирующим цепь ДНК на РНК-матрице (см. разд. 5.5.8). Одноцепочечные молекулы ДНК, синтезированные обратной транскриптазой, превращаются в двухцепочечные молекулы ДНК под действием ДНК-полимеразы, а эти двухцепочечные молекулы включаются в плазмиды и образуют клоны (рис. 5-80). Каждый полученный таким путем клон называется клоном кДНК, а вся совокупность клонов, происходящих от одного препарата мРНК, составляет библиотеку кДНК.
Как видно из рис. 5-81, клоны геномной ДНК и клоны кДНК существенным образом различаются. Геномные клоны представляют собой случайную выборку из всех нуклеотидных последовательностей ДНК данного организма. Состав геномной библиотеки не зависит от того, какой тип клеток был выбран для ее получения. В отличие от этого клоны кДНК содержат лишь те участки генома, которые транскрибируются в мРНК, а так как в клетках разных тканей наборы синтезируемых молекул мРНК различны, то и библиотеки кДНК различны для разных типов клеток, используемых при их конструировании.
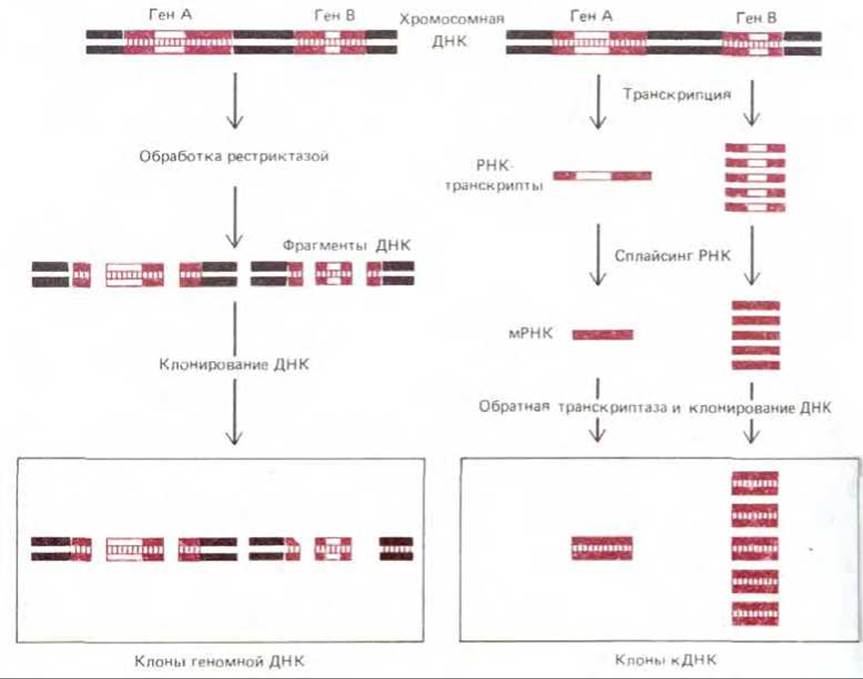
Рис. 5-81. Схема, иллюстрирующая различия между клонами кДНК и клонами геномной ДНК. В этом примере ген А транскрибируется редко, а ген В - часто. Оба типа РНК-транскриптов подвергаются процессингу путем сплайсинга, в результате чего при образовании мРНК удаляется единственный представленный здесь интрон. Большинство генов содержит много интронов (см. табл. 9-1).
Клонирование генов на основе библиотек кДНК имеет ряд преимуществ. Первое из них связано с тем, что многие белки вырабатываются специализированными клетками в очень больших количествах, а значит, в избытке синтезируется и мРНК, кодирующая подобный белок. Библиотека кДНК для таких клеток, естественно, обогащена молекулами кДНК, кодирующей данный белок (рис. 5-81). Это обилие именно нужных молекул кДНК сильно облегчает задачу распознавания требуемого клона в библиотеке. Гемоглобин, например, синтезируется в больших количествах в развивающихся эритроцитах, и как раз по этой причине в числе первых подвергнутых клонированию генов были гены глобина.
Другое преимущество клонов кДНК заключается в том, что в них кодирующая нуклеотидная последовательность гена ничем не прерывается. Эукариотические гены, как известно, состоят из кодирующих последовательностей ДНК, разделенных некодирующими, так что синтез мРНК на таких генах должен сопровождаться удалением из исходного РНК-транскрипта некодирующих (интронных) участков и сплайсингом (сращиванием) остающихся фрагментов, т. е. кодирующих последовательностей. Ни бактериальные, ни дрожжевые клетки не способны осуществить такую модификацию РНК, образовавшейся путем транскрипции гена высшей эукариотической клетки. Поэтому, если цель клонирования состоит в определении аминокислотной последовательности белка по данной ДНК или в получении больших количеств белка путем экспрессии клонированного гена в бактериальной или дрожжевой клетке, то начинать предпочтительнее именно с кДНК.
Библиотеки геномной ДНК и к ДНК - неисчерпаемый источник для соответствующих работ и исследователи часто пользуются ими сообща; неуклонно растет также и число таких библиотек, поставляемых специализированными фирмами.
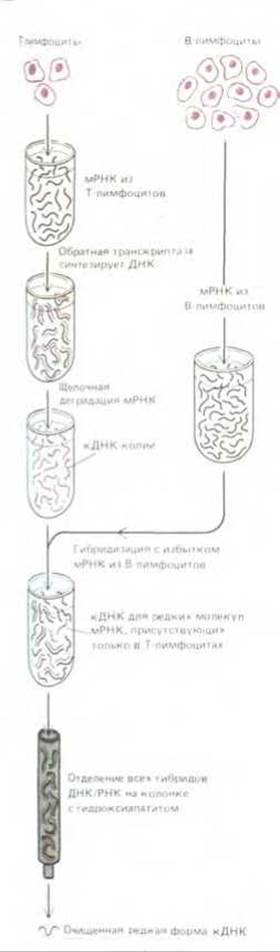
Рис. 5-82. Применение субтрактивной гибридизации для очистки тех редких клонов кДНК, которые соответствуют молекулам мРНК, имеющимся в Т-лимфоцитах, но отсутствующих в В-лимфоцитах. Поскольку два этих типа клеток очень сходны, большая часть мРНК должна быть свойственна как тому, так и другому типу. Данная процедура, таким образом, представляет собой очень эффективный способ обогащения теми специализированными молекулами, по которым два этих типа клеток отличаются друг от друга.
10-5
5.6.4. Библиотеки кДНК могут быть получены из отобранных популяций молекул мРНК [58]
Если кДНК получают из клеток, в которых уровень экспрессии интересующего нас гена очень высок, то большинство клонов кДНК содержит нуклеотидную последовательность этого гена, и, значит, отбор его не представляет трудностей. Для генов, транскрибируемых не столь интенсивно, необходимы какие-то приемы, которые позволили бы обогатить имеющуюся смесь молекул мРНК нужным ее видом и лишь после этого приступать к получению библиотеки кДНК. Если, например, в нашем распоряжении имеются антитела к данному белку, то ими можно воспользоваться для избирательного осаждения тех изолированных полирибосом, которые содержат его растущие полипептидные цепи. В этих рибосомах будет находиться также и мРНК, кодирующая данный белок, вследствие чего преципитат может быть обогащен интересующей нас мРНК иногда в 1000 раз.
Субтрактивная гибридизация представляет собой еще один высокоэффективный метод для обогащения смеси молекул определенными нуклеотидными последовательностями перед клонированием кДНК. Эту процедуру отбора можно использовать, например, в том случае, если имеются два типа клеток от одного и того же вида организма, очень близких, но отличающихся тем, что лишь один из них продуцирует интересующий нас белок (или белки). Впервые этот метод был применен для идентификации рецепторных белков, имеющихся на поверхности у Т- лимфоцитов, но отсутствующих у В-лимфоцитов. Его можно использовать и в том случае, если у клетки, вырабатывающей определенный белок, имеется мутантный двойник, лишенный такой способности. На первом этапе требуется осуществить синтез кДНК, используя для этого мРНК из того типа клеток, который вырабатывает нужный белок. Затем проводят гибридизацию этих кДНК с молекулами мРНК из клеток второго типа, добавленными в большом избытке. При этом небольшая часть нуклеотидных последовательностей кДНК не находит себе партнера, т.е. комплементарной последовательности мРНК. Очевидно, что именно эти немногие последовательности кДНК соответствуют последовательностям мРНК, имеющимся только у клеток первого типа. Их можно подвергнуть очистке, воспользовавшись для этого простой биохимической процедурой, дающей возможность отделить одноцепочечные нуклеиновые кислоты от двухцепочечных (рис. 5-82). Библиотеки кДНК, полученные путем субтрактивной гибридизации, удобны не только для клонирования тех генов, продукты которых приурочены к определенному типу дифференцированных клеток; они позволяют также определять различия в экспрессии генов между любыми двумя близкими типами клеток.
5-52
5.6.5. Для выявления нужных клонов в генной библиотеке можно использовать гибридизацию с радиоактивным ДНК-зондом [59]
При клонировании генов самой трудной задачей является распознавание в библиотеке тех редких колоний, которые содержат интересующий нас фрагмент ДНК. Это особенно справедливо в отношении геномной библиотеки, так как здесь, для того чтобы выделить нужный ген млекопитающего, приходится среди миллиона клеток разыскивать какую-нибудь одну. Чаще всего для этой цели пользуются особой формой гибридизации in situ, основанной на высокой специфичности
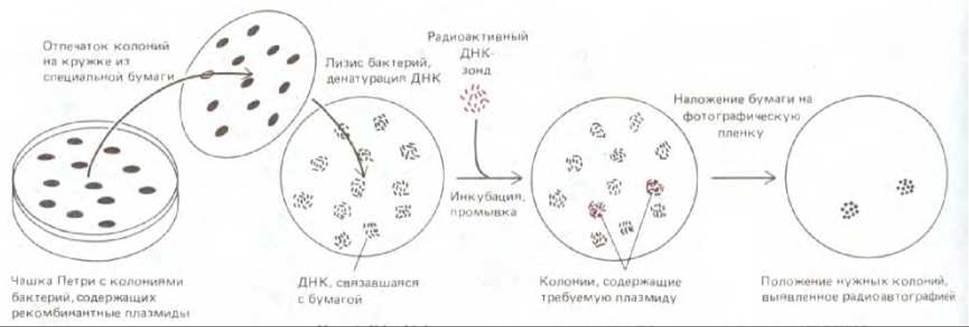
Рис. 5-83. Эффективный метод, широко применяемый для выявления бактериальной колонии, содержащей определенный клон ДНК (см. также рис. 5-79). Каждая бактериальная клетка, несущая рекомбинантную плазмиду, дает начало колонии из идентичных клеток, которая на питательном агаре выглядит как белое пятнышко. Прижимая к поверхности чашки кружок из фильтровальной бумаги, получают реплику бактериальной культуры. Эту реплику обрабатывают щелочью, чтобы разрушить прилипшие к бумаге клетки и денатурировать плазмидную ДНК, а затем проводят гибридизацию с высокорадиоактивным ДНК-зондом. Колонии бактерий, связавшие ДНК-зонд, выявляют методом радиоавтографии.
комплементарных взаимодействий между двумя комплементарными молекулами нуклеиновых кислот. Растущие на чашках колонии бактерий промакивают куском фильтровальной бумаги. При этом часть клеток от каждой колонии прилипает к бумаге. Это прилипшие колонии - их называют репликами - обрабатывают щелочью, а затем инкубируют с радиоактивным ДНК-зондом, содержащим часть нуклеотидной последовательности искомого гена (рис. 5-83). При необходимости такому скринингу можно подвергнуть миллионы бактериальных клонов, для того чтобы выявить тот клон, который способен к гибридизации с данным зондом.

Рис. 5-84. Выбор участков с известной аминокислотной последовательностью для приготовления синтетических олигонуклеотидных зондов. В действительности кодирует данный белок только одна какая-то нуклеотидная последовательность. Однако вследствие вырожденности генетического кода несколько разных нуклеотидных последовательностей могут дать одну и ту же аминокислотную последовательность, так что нельзя заранее сказать, какая из них окажется правильной. Желательно, чтобы в смеси олигонуклеотидов, используемых в качестве зонда, правильная нуклеотидная последовательность составляла наибольшую фракцию, поэтому выбирают участки, для которых число возможностей минимально, как это видно на рисунке. После того как смесь олигонуклеотидов будет синтезирована химическим путем, 5'-конец каждого олигонуклеотида метят радиоактивной меткой (см. рис. 4-65, Б).
Способ получения специфического ДНК-зонда зависит от той информации, которой мы располагаем в отношении клонируемого гена. Во многих случаях интересующий нас белок можно идентифицировать химическими методами и выделить хотя бы в малых количествах в очищенном виде. Нескольких микрограмм чистого белка достаточно для того, чтобы определить последовательность первых трех десятков аминокислот в его молекуле. Из этой аминокислотной последовательности на основе генетического кода можно вывести соответствующую нуклеотидную последовательность. Получив эти данные, синтезируют химическим путем два набора олигонуклеотидов ДНК, каждый длиной приблизительно 20 нуклеотидов, и вводят в них радиоактивную метку. Два набора используют в расчете обеспечить соответствие двум разным участкам предсказанной нуклеотидной последовательности клонируемого гена (рис. 5-84). Колонии клеток, обнаружившие способность к гибридизации с обоими наборами ДНК-зондов, скорее всего содержат требуемый ген; поэтому именно их сохраняют для дальнейшего исследования (см. ниже).
5.6.6. Выделение перекрывающихся клонов ДНК («прогулка по хромосоме») позволяет идентифицировать ген, находящийся по соседству с тем, который уже клонирован [60]
Многие гены из числа тех, что представляют наибольший интерес (например гены, регулирующие развитие), известны нам только из генетического анализа мутантов у таких организмов, как плодовая мушка Drosophila или нематода С. elegans. Белковые продукты этих генов не выделены; возможно, что они присутствуют лишь на какой-то определенной стадии развития. Тем не менее, изучая генетическое сцепление различных мутаций, можно составлять хромосомные карты, дающие представление об относительном расположении этих генов. Если один из картированных таким образом генов удалось клонировать, то клоны в библиотеке геномной ДНК, соответствующие соседним генам, можно идентифицировать при помощи методики, известной как «прогулка по хромосоме». Для этого используют две разные библиотеки геномной ДНК, полученные из одной и той же ДНК путем разделения ее на фрагменты двумя разными рестрицирующими нуклеазами. Рис. 5-85 показывает, как клон одной из библиотек может служить ДНК-зондом для выявления перекрывающегося клона другой библиотеки. Из этого нового клона получают затем ДНК-зонд, используемый для нахождения другого перекрывающегося клона в первой библиотеке, и т.д. Таким путем, отыскивая клон за клоном, можно продвигаться по хромосоме всякий раз на расстояние порядка 30000 пар нуклеотидов и более. Как узнать, однако, когда мы, наконец, дойдем до интересующего нас гена (идентифицированного изначально по какой-нибудь вредной мутации)? Обычный прием состоит в том, чтобы непрерывно сравнивать размеры рестрикционных фрагментов ДНК мутантных и нормальных хромосом посредством блот-анализа по Саузерну, используя в процессе «прогулки» в качестве зонда каждый новый клон. Некоторые из мутантов возникают вследствие небольших делеций или, наоборот, вставок последовательностей ДНК в соответствующем гене, и определить такие нарушения, как правило, бывает нетрудно. Известно, например, что среди вредных мутаций, затрагивающих типичный ген человека, примерно одна из десяти представляет собой делецию, легко обнаруживаемую блот-анализом по Саузерну. Число выявленных таким путем молекулярных дефектов, ответственных за наследственные болезни человека, в последнее время непрерывно растет.
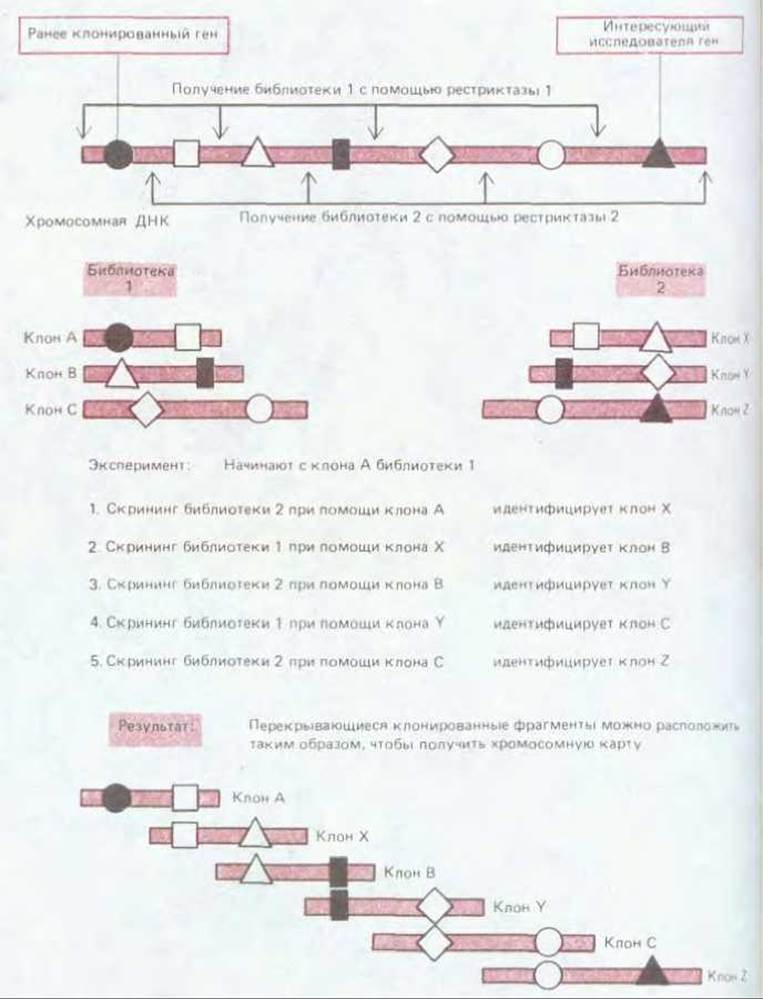
Рис. 5-85. Использование перекрывающихся фрагментов для картирования интересующего нас гена путем «прогулки по хромосоме». Для того чтобы сократить время «прогулки», наиболее пригодны геномные библиотеки, содержащие очень крупные клонированные молекулы ДНК. Зондом для каждого следующего клона служит короткий 32Р-фрагмент ДНК одного из концов предыдущего идентифицированного клона. Если, например, используется «правый» конец, то и перемещение происходит «вправо», как в случае, представленном на этом рисунке. Короткий концевой фрагмент удобен в качестве зонда еще и потому, что это снижает вероятность присутствия в зонде повторяющейся последовательности ДНК, которая могла бы гибридизоваться со многими клонами из разных частей генома и тем самым прервать «прогулку».
С помощью сходных методов можно расположить в правильном порядке (картировать) в хромосомах С. elegans почти весь набор крупных геномных клонов. Такие крупные клоны, каждый примерно по 30000 п.н., вводят в специальные векторы, приготовленные на основе фага λ, так называемые космиды, пригодные для включения больших вставок ДНК. Нескольких тысяч космидных клонов достаточно для того, чтобы охватить весь геном такого организма, как С. elegans или Drosophila, а чтобы картировать таким способом геном человека, необходимо более 100000 космидных клонов. Эта процедура заняла бы, конечно, очень много времени, но технически она выполнима. Кроме того, фрагменты ДНК человека, в 10 раз более крупные, чем космидные клоны (300000 п. н.), можно клонировать, как искусственные хромосомы в дрожжевых клетках; в принципе геном человека можно было бы картировать с помощью 10000 клонов такого типа (см. рис. 9-5).
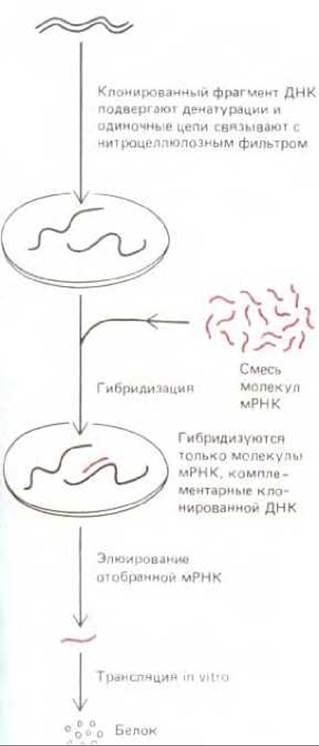
Рис. 5-86. Методика гибридизационной селекции. Молекулы очищенной мРНК элюируются с фильтра в условиях, вызывающих разделение РНК-ДНК-спирали на одиночные цепи.
В недалеком будущем исследователи, несомненно, получат возможность приобретать систематизированные наборы геномных клонов в специальных центрах, располагающих библиотеками ДНК. Там можно будет получить полную библиотеку для всякого обычного объекта исследования с указанием в каталогах для каждой вставки ДНК как хромосомы, из которой она взята, так и ее порядкового номера относительно всех других фрагментов ДНК, происходящих из той же хромосомы. Тогда начать «прогулку по хромосоме» можно будет просто, получив из библиотеки все клоны, охватывающие тот ее участок, в котором содержится интересующий исследователя мутантный ген. Эти клоны послужат затем для приготовления ДНК-зондов, которые позволят точно локализовать измененный ген. В конце концов таким путем удастся выделить многие из мутантных генов, обусловливающих наследственные заболевания у человека.
5.6.7. Трансляция in vitro облегчает идентификацию надлежащего клона ДНК [61]
Выделение клонов геномной ДНК и кДНК методом гибридизации in situ при всей своей эффективности не вполне удобно тем, что в таких экспериментах отбирается обычно немало и «псевдоположительных» клонов. Требуются дополнительные приемы для того, чтобы отличить их от подлинных. Задача облегчается, если требуемый клон кодирует какой-нибудь белок, ранее уже охарактеризованный иными методами. В этом случае каждый подлежащий проверке клонированный фрагмент ДНК используют для отбора комплементарных ему молекул мРНК из смеси клеточных мРНК посредством процесса, называемого гибридизационной селекцией; при этом избыток данного фрагмента ДНК расщепляют на одиночные цепи, иммобилизуют на фильтре и наносят на этот фильтр смесь мРНК, чтобы отобрать комплементарные молекулы мРНК путем РНК- ДНК-гибридизации (рис. 5-86). Очищенную таким способом мРНК используют затем для синтеза белка в бесклеточной системе, содержащей радиоактивные аминокислоты. Полученный радиоактивный белок исследуют и сравнивают с белковым продуктом, который, согласно ожиданиям, должен быть получен от данного клона. Совпадение их характеристик служит обычно в таком тесте основанием для вывода, что клонируемый фрагмент ДНК кодирует данный белок.
5.6.8. Экспрессирующие векторы могут быть использованы для сверхпродукции белков [62]
Очень часто никаких биохимических данных относительно белка, кодируемого тем или иным клонированным фрагментом ДНК, не имеется. Именно так, например, обстоит дело в тех случаях, когда клон, о котором идет речь, был идентифицирован посредством субтрактивной гибридизации или же в результате «прогулки по хромосоме» до соответствующего мутантного гена. Более того, в таких случаях мРНК, кодирующая этот белок, часто присутствует в столь малых количествах или же в столь немногих клетках, что гибридизационная селекция комплементарной мРНК оказывается неосуществимой. При этом приходится прибегать к другим методам, которые дают возможность охарактеризовать белковый продукт клонированного гена. Один из методов состоит в том, чтобы синтезировать короткий фрагмент белка (олигопептид), соответствующий выведенной аминокислотной последовательности белкового продукта секвенированной молекулы кДНК, а затем получить антитела к этому олигопептиду. Эти антитела во многих случаях будут распознавать ту же аминокислотную последовательность в составе природного белка, что даст возможность обнаруживать, локализовать и подвергать очистке белок, кодируемый исходной кДНК. Такой иммунологический подход в сочетании с клонированием путем субтрактивной гибридизации эффективен и как способ идентификации белков, специфичных для определенного типа клеток, и как путь к изучению характера дифференцировки, а также свойств и функций каждого типа клеток в многоклеточном организме.
Однако самый простой путь, позволяющий охарактеризовать белок, закодированный в какой-нибудь клонированной кДНК, заключается в том, чтобы заставить саму эту кДНК направлять синтез белка в клетке-хозяине. Плазмиды или вирусы выступают в таких случаях в качестве экспрессирующих векторов; их конструируют с таким расчетом, чтобы присоединить клонированную кДНК к той последовательности, которая служит сильным промотором для транскрипции. Исследователи располагают достаточно широким набором экспрессирующих векторов - каждый из них приспособлен для функционирования в клетках того типа, в которых должен синтезироваться данный белок. С помощью этого генноинженерного подхода клетки бактерий, дрожжей и млекопитающих можно заставить вырабатывать большие количества различных ценных белков, таких, как гормон роста человека, интерферон или антитела к вирусам, используемые для приготовления вакцин. Особенно удобны для получения белка бактериальные клетки с плазмидными или вирусными векторами, сконструированными подобным образом; встроенный чужеродный ген обеспечивает нередко свыше 10% от всего синтезируемого клеткой белка. Для клетки синтез такого большого количества какого-нибудь белка оказывается часто непомерной метаболической нагрузкой; поэтому предложены особые «индуцибельные» промоторы, с помощью которых запуск транскрипции можно осуществлять лишь за несколько часов до сбора клеток для выделения из них белка. Некоторые плазмидные экспрессирующие векторы содержат, например, промотор, полученный из бактериофага X. Работа его регулируется температурочувствительным белковым продуктом генарепрессора; повысив температуру бактериальных клеток до 42°С, можно в любой момент «включить» промотор и быстро получить большое количество требуемого белка.
Если библиотека кДНК создана в экспрессирующем векторе, то каждый клон будет продуцировать особый белок и тогда представляющие интерес клоны можно идентифицировать не по их нуклеотидной последовательности, а по их белковым продуктам. При этом в качестве зонда для клонов обычно применяют радиоактивно меченные антитела. Иногда вместо этого можно провести прямой тест, выявляющий биологическую активность продукта данного гена. Особенно эффективен этот метод при поисках эукариотических генов, ответственных за секретируемые факторы роста. Можно, например, клоны кДНК из клеток, вырабатывающих определенный фактор роста, ввести в экспрессирующий вектор, размножающийся в клетках млекопитающих. Смесь трансфицированных клеток, содержащих много таких различных клонов, выращивают на небольшой чашке, где каждая клетка, в которой экспрессируется данный ген, выделяет этот фактор роста в среду. Затем пробы среды испытывают на присутствие данного фактора роста, добавляя их к культурам других клеток, способных реагировать на этот фактор. Тест отличается настолько высокой чувствительностью, что положительный ответ получают даже в том случае, когда в исходной культуре одна клетка из тысячи содержит ген, кодирующий данный фактор роста. Повторное тестирование позволяет отыскать в смеси тот единственный клон, который продуцирует этот фактор. Таким способом удалось открыть ранее неизвестные факторы роста и выделить их за срок, не превышавший нескольких месяцев; если бы процедуру вели обычными биохимическими методами, то для очистки в миллион с лишним раз потребовалась бы чрезвычайно кропотливая работа, которая длилась бы годы.
5-53
5.6.9. Гены можно «перестраивать» и таким путем получать белки с желаемой аминокислотной последовательностью [63]
Перестраивая кодирующую последовательность и регуляторные участки гена, можно изменять функциональные свойства его белкового продукта, количество синтезируемого белка и, наконец, менять тип клеток, способных вырабатывать данный белок.
В кодирующую последовательность можно вносить весьма существенные изменения, например, можно «пришивать» какую-нибудь ее часть к другому гену, что даст в результате новый гибридный ген, кодирующий комбинированный (составной) белок (fusion protein). Подобные белки часто используются для выявления функций различных доменов белковой молекулы. Известно, например, что большинство ядерных белков содержит особые короткие аминокислотные последовательности, которые распознаются как сигнал для немедленного импорта этих белков в клеточное ядро. Присоединяя искусственно - методом слияния генов - к какому-нибудь цитоплазматическому белку различные части молекулы ядерных белков, можно идентифицировать эти «сигнальные пептиды», ответственные за импорт в ядро.
Для более тонкой структурной реорганизации гена (результатом которой является замена одной или нескольких аминокислот в кодируемом белке) требуются специальные методы. Сначала химическим путем синтезируют небольшую молекулу ДНК, содержащую измененную часть нуклеотидной последовательности данного гена. Затем этот синтетический олигонуклеотид гибридизуют с одноцепочечной плазмидой ДНК, в составе которой присутствует последовательность ДНК, подлежащая изменению; гибридизацию ведут в условиях, допускающих спаривание не вполне подходящих партнеров (рис. 5-87). Синтетический олигонуклеотид становится затравкой для ДНК-полимеразы, осуществляющей синтез ДНК, в результате которого образуется молекула, содержащая ген с встроенной в него измененной последовательностью. После этого модифицированный ген включают в экспрессирующий вектор, что позволяет получить измененный белок в достаточном количестве для детального изучения. Заменяя таким путем те или иные аминокислоты в молекуле интересующего нас белка, можно выяснить, какие именно части полипептидной цепи ответственны за такие фундаментальные процессы, как свертывание белка, взаимодействие белка с лигандом или ферментативный катализ.
В тех случаях, когда требуется определить, какая именно часть эукариотического гена ответственна за регулирование его экспрессии, предполагаемые регуляторные последовательности ДНК этого гена можно присоединить к кодирующей последовательности другого гена, выбранного с таким расчетом, чтобы в нем был закодирован какой-нибудь легко обнаруживаемый фермент-маркер, в норме в эукариотических клетках отсутствующий. Чаще всего в качестве такого фермента-маркера используется бактериальный белок хлорамфеникол-ацетил-трансфераза (ХАТ). Полученную рекомбинантную молекулу ДНК вводят в эукариотическую клетку, чтобы выяснить, действительно ли предполагаемые регуляторные последовательности стимулируют экспрессию гена ХАТ. Проще всего сделать это, если трансфицировать клетки в культуре и затем после инкубации в течение ночи исследовать их на хлорамфеникол-ацетилтрансферазную активность. Среди трансфицированных клеток многие в течение некоторого времени экспрессируют чужеродный ген, но лишь очень небольшое число клеток удерживает этот ген надолго. Чтобы добиться устойчивой трансформации, надо произвести отбор тех редких клеточных клонов, у которых рекомбинантные молекулы ДНК включились в их хромосомы. Трансфицируя различные типы клеток такими рекомбинантными молекулами ДНК, удается определять те регуляторные последовательности ДНК, которые позволяют данному гену экспрессироваться в одних клетках и не экспрессироваться в других (разд. 10.2.8).
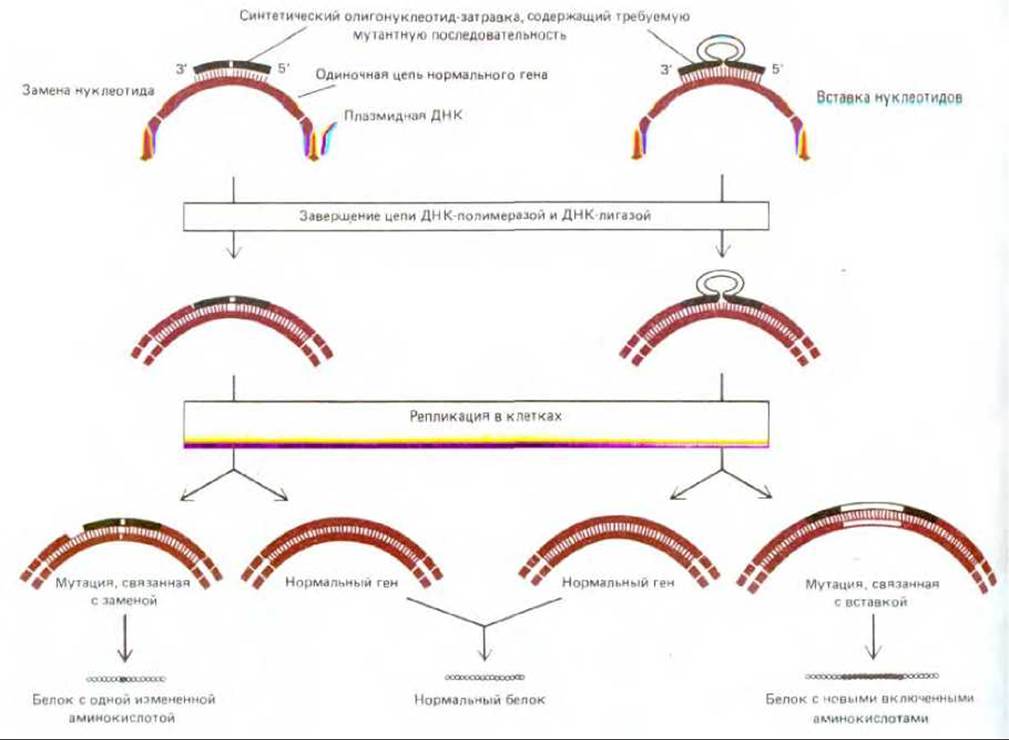
Рис. 5-87. Использование синтетических олигонуклеотидов для изменения отдельных кодирующих участков гена. Здесь приведены лишь два примера из многих возможных типов таких изменений. С помощью подходящего олигонуклеотида можно, например, заменить одновременно не одну, а несколько аминокислот, можно также вызвать выпадение одной или нескольких аминокислот. Из схемы ясно, что при такой процедуре в исходной рекомбинантной плазмиде изменяется только одна из двух цепей ДНК, так что после репликации лишь половина всех трансфицированных клеток содержит плазмиду с нужным мутантным геном. Здесь указана лишь небольшая часть всей плазмидной последовательности.
5.6.10. Сконструированные гены можно ввести в половые клетки мыши или плодовой мушки и получить трансгенные организмы [64]
Для выявления функции измененного белка, необходимо ввести этот ген в организм и проследить, каков будет эффект. В идеале желательно заменить нормальный ген измененным, с тем чтобы действие мутантного белка можно было наблюдать в условиях, когда нормальный белок отсутствует. Единственный эукариотический организм, с которым такую процедуру можно проделать относительно легко, - это дрожжи. Фрагменты ДНК, введенные в растущие дрожжевые клетки, достаточно эффективно включаются в виде одиночных копий в гомологичные участки хромосом путем общей рекомбинации, так что на место эндогенной копии данного гена становится сконструированный ген. Это
открывает широкие возможности для изучения функций различных генов у дрожжей (см. разд. 4.6.15).
Иначе обстоит дело с клетками млекопитающих. Здесь мы пока не в состоянии установить, как и куда включилась в хромосому введенная в клетку ДНК. В среднем лишь один акт интеграции на тысячу приводит к замене одного гена другим. Вместо этого клеточные ферменты быстро сшивают линейные фрагменты ДНК конец-в-конец и такой длинный тандем обычно включается в хромосому в каких-то, по-видимому, случайных, участках. Оплодотворенные яйца млекопитающих ведут себя в этом отношении так же, как и прочие клетки млекопитающих. Если ввести в яйцо мыши 200 копий линейной молекулы ДНК, то часто из него развивается мышь, у которой в одну из ее хромосом в случайном участке интегрированы эти тандемно повторяющиеся копии введенного гена (рис. 5-88). В том случае, когда модифицированная хромосома присутствует и в половых клетках (яйцеклетках или сперматозоидах), мышь передаст эти новые полученные ею гены своему потомству; животные с таким устойчивым изменением называются трансгенными. Поскольку нормальный ген у них, как правило, сохраняется, трансгенные животные по большей части непригодны для столь убедительной, как у дрожжей, демонстрации эффектов, связанных с изменением гена; однако с их помощью были уже получены важные сведения о том, как осуществляется регуляция генов млекопитающих (см. стр. 10.3.11) и каким образом некоторые гены (их называют онкогенами) обусловливают возникновение рака (см. разд. 21.2.5).
Сходные эксперименты можно проводить с плодовой мушкой Drosophila, используя для этого методику, дающую возможность включать в геном мухи в каком-либо случайном его участке одну копию любого интересующего нас гена. Главная задача состоит здесь в том, чтобы встроить фрагмент ДНК, о котором идет речь, между двумя концевыми последовательностями определенного мобильного элемента Drosophila, так называемого Р-элемента. Эти концевые последовательности позволяют Р-элементу включаться в хромосомы Drosophila, если в клетке присутствует также особый фермент, катализирующий это включение (Р-элемент-транспозаза; см. разд. 5.5.10). Поэтому, для того чтобы получить трансгенных плодовых мушек, требуется инъецировать соответствующим образом модифицированный фрагмент ДНК в эмбрион мухи на одной из очень ранних стадий развития и одновременно ввести туда же отдельную плазмиду, содержащую ген транспозазы. В этом случае инъецированный ген в виде одиночной копии часто попадает (вследствие транспозиции) в клетки зародышевого пути (рис. 5-88).
Недавно у мышей удалось осуществить замену гена косвенным, достаточно трудоемким путем. Процедура сводится к следующему. Сначала фрагмент ДНК, содержащий нужный мутантный ген, вводят путем трансфекции в специальную линию плюрипотентных стволовых клеток, выделенных из эмбриона и выращиваемых в культуре. Клеткам дают возможность размножаться в течение некоторого времени, после чего методом блот-анализа по Саузерну выявляют редкие колонии, в которых общая рекомбинация привела к замене гена, и отдельные клетки из этих колоний имплантируют в ранний эмбрион мыши. В этой новой для них среде выделенные из эмбриона стволовые клетки часто пролиферируют, образуя крупные участки в теле нормальной мыши. Мышей, у которых произошла замена гена в клетках зародышевого пути, скрещивают, чтобы получить самца и самку, которые окажутся гетерозиготными по этому признаку (т. е. будут иметь одну нормальную и одну мутантную копию данного гена). Если теперь скрестить этих животных, то одна четвертая часть их потомства будет гомозиготной по измененному гену. Изучение таких гомозигот позволит наблюдать функцию измененного гена в отсутствие его нормального аллеля.
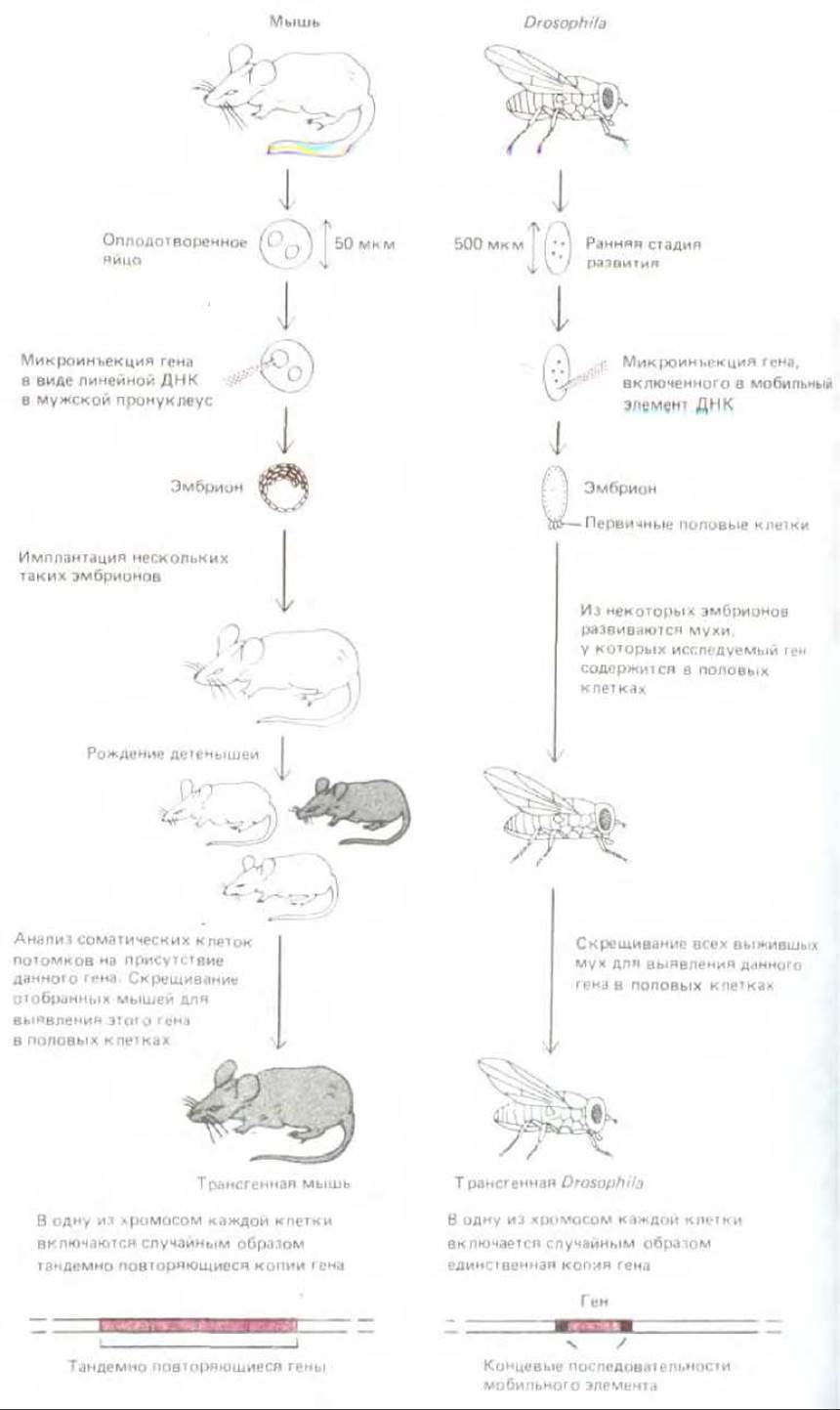
Рис. 5-88. Сравнение стандартных процедур, используемых для получения трансгенной мыши и трансгенной плодовой мушки. В этих примерах ген, введенный в яйцо мыши, вызывает изменение окраски шерсти, а ген, введенный в эмбрион плодовой мушки, - изменение цвета глаз. В обоих случаях у части трансгенных организмов вставки ДНК обнаружены не в одном, а в нескольких участках хромосомы.
Возможность получения трансгенных мышей и дрозофил дает в руки исследователей новый мощный инструмент для изучения функции генов в интактном организме.
5.6.11. Отобранные сегменты ДНК можно клонировать in vitro посредством полимеразной цепной реакции [65]
Доступность очищенных ДНК-полимераз и химически синтезированных ДНК-олигонуклеотидов сделала возможным быстрое клонирование специфических последовательностей ДНК вне живой клетки. Методика, известная под названием полимеразной цепной реакции (ПЦР), позволяет амплифицировать ДНК из какого-нибудь выбранного участка генома более чем в миллион раз, при условии, что нам известна хотя бы часть его нуклеотидной последовательности. Участки этой последовательности, окружающие выбранную для амплификации область, используют для того, чтобы приготовить по ним два синтетических ДНК-олигонуклеотида, каждый из которых комплементарен одной из двух цепей молекулы ДНК. Эти олигонуклеотиды служат затравками при синтезе ДНК in vitro катализируемом ДНК-полимеразой; они определяют концы получаемого фрагмента ДНК (рис. 5-89, А).
Принцип методики ПЦР ясен из рис. 5-89, Б. В каждом цикле реакции необходимо сначала кратковременное нагревание ДНК для разделения двух цепей двойной спирали (1-й этап). Последующее охлаждение ДНК в присутствии большого избытка двух упомянутых ДНК- олигонуклеотидов приводит к специфической гибридизации этих олигонуклеотидов с комплементарными последовательностями ДНК (2-й этап). После отжига смесь инкубируют с ДНК-полимеразой и четырьмя дезоксинуклеозидтрифосфатами, в результате чего избирательно синтезируются те участки ДНК, которые располагаются «книзу» от затравки (3-й этап). Для эффективной амплификации ДНК требуется от 20 до 30 циклов реакции. В каждом последующем цикле количество ДНК по сравнению с предыдущим циклом удваивается. Отдельный цикл занимает около 5 мин, поэтому при автоматизации всей процедуры для «бесклеточного молекулярного клонирования» какого-либо фрагмента ДНК требуется несколько часов, тогда как обычные процедуры клонирования растягиваются на несколько дней.
Метод ПЦР отличается чрезвычайно высокой чувствительностью: он позволяет обнаружить в пробе всего одну присутствующую в ней молекулу ДНК. Тот же способ пригоден и для анализа следовых количеств РНК; для этого РНК переводят в последовательности ДНК с помощью обратной транскриптазы (см. разд. 5.6.3). Клонирование методом ПЦР быстро вытесняет блот-анализ по Саузерну в таких областях, как пренатальная диагностика наследственных болезней и выявление вирусных инфекций. Большие перспективы открывает этот метод также и в судебной медицине, поскольку он дает возможность однозначно определить принадлежность одной-единственной клетки данному человеку.
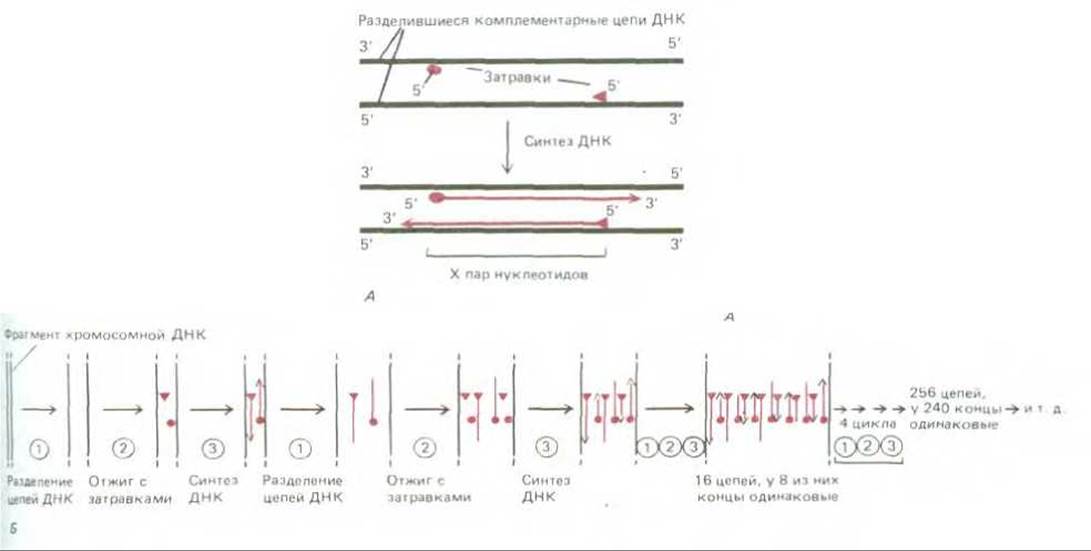
Рис. 5-89. Полимеразная цепная реакция (ПЦР), используемая для амплификации специфических нуклеотидных последовательностей in vitro А. Выделенную из клеток ДНК нагревают, чтобы разделить ее комплементарные цепи. Затем проводят отжиг этих цепей, добавив к ним избыток двух ДНК-олигонуклеотидов (по 15-20 нуклеотидов каждый), синтезированных химически с таким расчетом, чтобы они были комплементарны последовательностям, отделенным друг от друга расстоянием в X нуклеотидов (значение X варьирует от 50 до 2000). Эти два олигонуклеотида служат специфическими затравками для синтеза ДНК in vitro, катализируемого ДНК-полимеразой, которая копирует ДНК между соответствующими последовательностями. Б. Многократное повторение циклов реакции дает в итоге большое количество копий одного фрагмента ДНК (длиной X нуклеотидов).
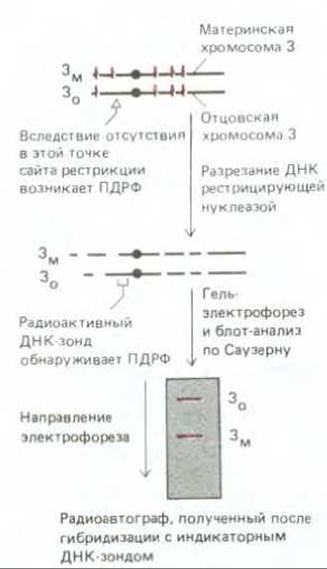
Рис. 5-90. Выявление полиморфизма длины рестрикционных фрагментов (ПДРФ) блот-анализом по Саузерну. Для простоты в хромосомах показано лишь несколько сайтов рестрикции, хотя в действительности их многие тысячи. Если до воздействия рестрицирующей нуклеазой провести амплификацию соответствующего участка методом ПЦР, этот же тест можно провести без радиоизотопов и от блот-анализа отказаться.
10-31
5.6.12. Применение рекомбинантных ДНК сильно облегчает картирование и анализ крупных геномов [66]
Появившиеся в последнее время методы позволяют составлять подробные карты очень больших геномов. Есть две категории карт: 1. Физические карты, основывающиеся на строении молекул ДНК, составляющих каждую хромосому. Сюда относятся рестрикционные карты и систематизированные библиотеки клонов геномной ДНК. 2. Карты генетического сцепления; их строят, основываясь на частоте совместной передачи потомству двух или нескольких признаков - генетических маркеров, различных у отца и матери и приписываемых определенному участку хромосомы. В качестве маркеров издавна принято использовать те гены, экспрессия которых обнаруживается по их эффекту (таковы, в частности, гены, вызывающие генетические болезни, например мышечную дистрофию). Разработанные сравнительно недавно новые методы с применением рекомбинантной ДНК дали возможность использовать в качестве генетических маркеров короткие последовательности ДНК, содержащие один из сайтов рестрикции и различающиеся у отдельных индивидуумов; такие последовательности особенно удобны для генетического картирования, потому что под действием рестрикционной нуклеазы возникают фрагменты, различающиеся по своей длине, и этот полиморфизм длины рестрикционных фрагментов (ПДРФ) легко может быть выявлен блот-анализом по Саузерну с помощью подходящего ДНК-зонда (рис. 5-90).
Если два генетических маркера находятся в разных хромосомах, то сцепление между ними отсутствует, т. е. шансы на их совместную передачу потомству равны 50:50. То же справедливо и в отношении маркеров, локализующихся на противоположных концах одной и той же хромосомы, из-за большой вероятности их разделения в результате кроссинговера, частота которого в процессе мейоза, при образовании яйцеклеток и сперматозоидов, весьма высока (см. разд. 15.2.3). Чем ближе друг к другу два маркера в пределах одной хромосомы, тем больше вероятность того, что они не будут разделены кроссинговером, а значит, будут переданы потомству совместно. Проведя скрининг больших семейных групп на совместное наследование интересующего нас гена (например, гена, ответственного за какую-нибудь болезнь) и большого числа отдельных ПДРФ-маркеров, можно идентифицировать несколько ПДРФ-маркеров, окружающих данный ген. Таким путем удается локализовать последовательности ДНК, находящиеся поблизости от этого гена, а в конце концов и ДНК, соответствующую самому этому гену (рис. 5-91). Этот метод используется для локализации многих генов, ответственных за болезни человека. После выделения такого гена можно подвергнуть детальному анализу его белковый продукт (см. разд. 4.6.12).
Чтобы облегчить эти и некоторые другие исследования, в настоящее время ведется усиленная работа по созданию детальной ПДРФ-карты генома человека, на которой будут отмечены тысячи маркеров, отстоящих друг от друга в среднем на 106 п. н. (два маркера, разделенные таким расстоянием, наследуются совместно в 99 случаях из 100). Получив в свое распоряжение такую карту, можно будет - на основе данных о генетическом сцеплении - гены, идентифицированные ранее лишь по их эффекту у человека, локализовать в пределах одного или нескольких больших клонов в систематизированной библиотеке геномной ДНК. После этого на выделение данного гена потребуется в большинстве случаев уже не более нескольких месяцев.
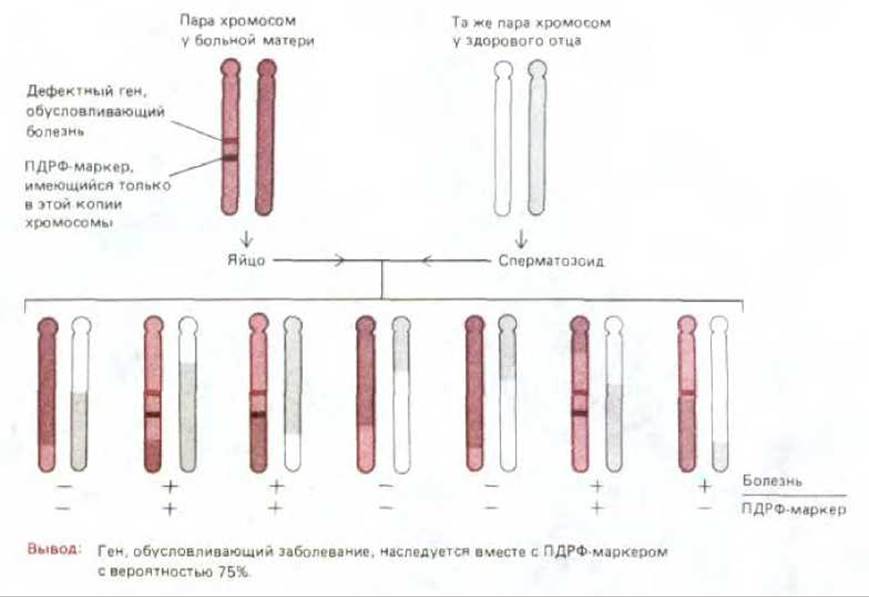
Рис. 5-91. Анализ генетического сцепления выявляет совместную передачу потомству какого-либо гена, обусловливающего у человека специфический фенотип (в данном случае - определенную болезнь), и соседнего ПДРФ-маркера. Такой анализ дает возможность клонировать многие гены человека и исследовать продукты этих генов.
Быстрые успехи технологии рекомбинантной ДНК позволят в ближайшее время секвенировать длинные отрезки ДНК при сравнительно небольших затратах. Это значит, что станет возможным секвенирование целых геномов бактерий, дрожжей, нематод и Drosophila, а также обширных участков генома различных высших растений и позвоночных животных, в том числе и человека.
Заключение
При клонированш ДНК фрагмент, содержащий изучаемый ген, выявляют обычно с помощью радиоактивного ДНК-зонда или, после экспрессии гена в клетке-хозяине, - с помощью антител, обнаруживающих кодируемый этим геном белок. Затем клеткам, несущим данный фрагмент ДНК, предоставляют возможность размножаться и нарабатывать большое количество копий как самого гена, так и молекул его продукта. Для генноинженерных задач нуклеотидную последовательность такого клонированного фрагмента ДНК изменяют, присоединяют к другой последовательности ДНК, а затем снова вводят в клетки. Сочетание клонирования ДНК с генной инженерией вооружает клеточного биолога очень мощным инструментом исследования. В принципе возможно сконструировать ген, кодирующий белок с любой желательной аминокислотной последовательностью, и присоединить его к такой промоторной последовательности ДНК, которая позволит контролировать время и тип экспрессии гена. Этот новый ген можно ввести либо в клетки, выращиваемые в культуре, либо в клетки зародышевого пути мыши или плодовой мушки. У трансгенных животных эффект экспрессии включенного гена можно наблюдать на многих различных клетках и тканях.
Литература
Общая
Judson И. F. The Eighth Day of Creation: Makers of the Revolution in Biology, New York, Simon and Schuster, 1979. (History derived from personal interviews.)
Lewin B. Genes, 3rd ed. New York, Wiley, 1987.
Stent G.S. Molecular Genetics: An Introductory Narrative, San Francisco, Freeman, 1971. (Clear descriptions of the classical experiments.) Watson J. D., Hopkins N. H.,
Roberts J. W., Weiner A. M. Molecular Biology of the Gene, 4th ed.Menlo Park, CA, Benjamin-Cummings 1987.
Цитируемая
1. Chamberlin M. Bacterial DNA-dependent RNA polymerases. In: The Enzymes, 3rd ed., Vol. 15B (P. Boyer ed.), pp. 61-108. New York, Academic Press, 1982.
McClure W. Mechanism and control of transcription initiation in prokaryotes.
Annu. Rev. Biochem., 54, 171-204, 1985.
Yager T.D., Hippel P.H. Transcript elongation and termination in E. coll In: Escherichia coli and Salmonella typhimurium: Cellular and Molecular Biology (F.C. Niedhardt ed.), pp. 1241-1275. Washington DC, American Society for Microbiology, 1987.
2. Hawley D. K., McClure W.R. Compilation and Analysis of Escherichia coli promoter DNA sequences. Nucleic Acids Res., 11, 2237-2255, 1983.
Siebenlist U., Simpson R., Gilbert W. E. coli RNA polymerase interacts homologously with two different promoters. Cell, 20, 269-281, 1980.
3. Rich A., Kim S. H. The three-dimensional structure of transfer RNA. Sci. Am, 238(1), 52-62, 1978.
Schimmel P. R., Soll D., Abelson J. N., eds. Transfer RNA: Structure, Properties and Recognition, and Biological Aspects (2 volumes). Cold Spring Harbor. NY, Cold Spring Harbor Laboratory, 1980.
4. Schimmel P. Aminoacyl tRNA synthetase: general scheme of structure-function relationships in the polypeptides and recognition of transfer RNAs. Annu. Rev. Biochem., 56, 125-158, 1987.
5. Crick F.H.C. The genetic code. III. Sci. Am., 21(4), 55-62, 1966. The Genetic Code. Cold Spring Harbor Symp. Quant. Biol., Vol. 31, 1966. (The original experiments that defined the code.)
6. Moore P. B. The ribosome returns. Nature, 331, 223-227, 1988.
Noller H.F. Structure of ribosomal RNA. Annu. Rev. Biochem., 53, 119-162, 1984.
Spirin A. S. Ribosome Structure and Protein Synthesis. Menlo Park CA, Benjamin-Cummings, 1986.
7. Clark B. The elongation step of protein biosynthesis. Trends Biochem. Sci., 5, 207-210, 1980.
Watson J. D. The involvement of RNA in the synthesis of proteins. Science, 140, 17-26, 1963. (A description of how ribosome function was initially deciphered.)
8. Caskey С. Т. Peptide chain termination. Trends Biochem. Sci., 5, 234-237, 1980. Cragien W.J., Caskey С. Т. The function, structure and
regulation of E. coli peptide chain release factors. Biochemie, 69, 1031-1041, 1987.
9. Gold L. Posttranscriptional regulatory mechanisms in Escherichia coli. Annu. Rev. Biochem., 57, 199-233, 1988.
Hunt T. The initiation of protein synthesis. Trends Biochem. Sci., 5, 178-181, 1980. Pain V.M. Initiation of protein synthesis in mammalian cells Biochem. J., 235, 625-637, 1986.
10. Kozak M. Comparison of initiation of protein synthesis in procaryotes, eucaryotes, and organelles. Microbiol. Rev., 47, 1-45, 1983.
Kozak M. An analysis of 5' noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res., 15, 8125-8147, 1987.
Steitz J. A. Genetic signals and nucleotide sequences in messenger RNA. In: Biological Regulation and Development (R. Goldberg ed.), Vol. 1, pp. 349-399. New York, Plenum, 1979.
11. Rich A. Polyribisomes. Sci. Am., 209(6), 44-53, 1963.
12. Hunt T. Phosphorylation and the control of protein synthesis. Philos. Trans. R. Soc. Lond. (Biol.), 302, 127-134, 1983.
Jagus R., Anderson W. F., Safer B. The regulation of initiation of mammalian protein synthesis. Prog. Nucleic Acid Res. Mol. Biol., 25, 127185, 1981.
13. Kirkwood T.B., Rosenberger R.F., Galas D. J., eds. Accuracy in Molecular Processes: Its Control and Relevance to Living Systems. London, Chapman and Hall, 1986. (Chapters 4, 5, 6 and 11.)
Thompson R. C. EFTu provides an internal kinetic standard for translational accurancy. Trends Biochem. Sci., 13, 91-93, 1988.
14. Jimenez A. Inhibibitors of translation. Trends Biochem. Sci., 1, 28-30, 1976.
15. Alberts B.M. The function of the hereditary materials: biological catalyses reflect the cell's evolutionary history. Am. Zool., 26, 781-796, 1986.
Orgel L.E. RNA catalysis and the origin of life. J. Theor. Biol. 123, 127-149, 1983.
Weiner A. M., Maizels N. tRNA-like structures tag the 3' ends of genomic RNA molecules for replication: implications for the origin of protein synthesis. Proc. Natl. Acad. Sci. USA, 84, 7383-7387, 1987.
16. Friedberg Е. С. DNA Repa ir. San Francisco, Freeman, 1985.
Lindahl T. DNA repair enzymes. Annu. Rev. Biochem., 51, 61-88, 1982.
17. Dickerson R., Geis 1. The Structure and Action of Proteins, pp. 59-66. New York, Harper and Row, 1969.
Wilson A. C., Carbon S. S., White T. J. Biochemical evolution. Annu. Rev. Biochem., 46, 573-639, 1977.
Wilson A. C., Ochman H., Prager Е. М. Molecular time scale for evolution. Trends Genet., 3, 241-247, 1987.
18. Drake J. M. Comparative rates of spontaneous mutation. Nature, 221, 1132, 1969.
19. Cairns J. Cancer: Science and Society. San Francisco. Freeman, 1978. (Chapter 7 discusses the role of mutation in the development of cancer.)
Ohta Т., Kimura M. Functional organization of genetic material as a product molecular evolution. Nature, 233, 118-119, 1971. (The mutation rate limits the maximum number of genes in an organism.)
20. Schrodinger E. What is Life? Cambridge UK, Cambridge University Press, 1945.
21. Kornberg A. DNA Replication. San Francisco, Freeman, 1980. (Chapter 4 describes DNA polymerase. Chapter 8 describes DNA ligase, and Chapter 16 covers the general enzymol ogy of DNA repair.)
22. Cleaver J. E., Karentz D. DNA repair in man: regulation by a multigene family and association with human disease. Bioessays, 6, 122-127, 1987.
Coulondre C., Miller J. H., Farabaugh P. J., Giobert W. Molecular basis of base substitution hotspots in E. coli. Nature, 274, 775-780, 1978. Lindahl T. New class of enzymes acting on damaged DNA. Nature, 259, 64-66, 1976.
Sancar A.Z., Sancar G.B. DNA repair enzymes. Annu. Rev. Biochem. 57, 29-67, 1988.
23. Lindahl Т., Sedgwick В., Sekiguchi M., Nakabeppu Y. Regulation and expression of the adaptive response to alkylating agents. Annu. Rev. Biochem., 57, 133-157, 1988.
Ossanna N.. Peterson K. R., Mount D. W. Genetics of DNA repair in bacteria. Trends Genet., 2, 55-58, 1986.
Walker G. C. Inducible DNA repair systems. Annu. Rev. Biochem., 54, 425-457, 1985.
24. Alberts B.M. Prokaryotic DNA replication mechanisms. Philos. Trans. R. Soc. bond. (Biol.), 317, 395-420, 1987.
Kornberg A. DNA Replication. San Francisco, Freeman, 1980.
McMacken R., Silver L., Georgopoulos C. DNA replication. In: Escherichia coli and Salmonella typhimurium: Cellular and Molecular Biology (F. C. Neidhardt ed.), pp. 564-612. Washington DC. American Society for Microbiology, 1987.
25. Joyce C.M., Steitz T.A. DNA polymerase I: from crystal structure to function via genetics. Trends Biocem. Sci., 12, 288-292, 1987.
Kornberg A. Biologic synthesis of deoxyribonuclear acid. Science, 131, 1503-1508. (Nobel prize address.)
Meselson M., Stahl F. W. The replication of DNA in E. coli. Proc. Natl. Acad. Sci. USA, 44, 671-682, 1958.
26. Cairns J. The bacterial chromosome and its manner of replication as seen by autoradiography. J. Mol. Biol., 6, 208-213, 1963.
Imman R. В., Schnos M. Structure of branch points in replicating DNA: presence of single-stranded connections in lambda DNA branch parts. J. Mol. Biol., 56, 319-325, 1971. (Electron microscopy demonstrates that the replication fork is asymmetric.)
Ogawa Т., Okazaki T. Discontinuous DNA replication. Annu. Rev. Biochem., 49, 421-457, 1980.
27. Brutlag D., Kornberg A. Enzymatic synthesis of deoxyribonucleic acid: a proofreading function for the 3' to 5' exonuclease activity in DNA polymerases. J. Biol. Chem., 247, 241-248, 1972.
Fersht A. R. Enzymatic editing mechanisms in protein synthesis and DNA replication. Trends Biochem. Sci., 5, 262-265, 1980.
Topal M., Fresco J. R. Complementary base pairing and the origin substitution mutations. Nature, 263, 285-289, 1976.
28. Rowen L., Kornberg A. Primase, the dnaG protein of Escherichia coli: an enzyme which starts DNA chains. J. Biol. Chem., 253, 758-764, 1978.
29. Abdel-Monem M., Hoffmann-Berling H. DNA unwinding enzymes. Trends Biochem. Sci., 5, 128-130, 1980.
Alberts В. М., Frey L. T4 bacteriophage gene 32: a structural protein in the replication and recombination of DNA, Nature, 227, 1313-1318, 1970.
Lohman T. M., Bujalowski W., Overman L. В. Е. coli single strand binding protein: a new look at helix-destabilizing proteins. Trends Biochem. Sci., 13, 250-254, 1988.
30. Alberts В. М. Protein machines mediate the basis genetic processes. Trends Genet., 1, 26-30, 1985.
Kornberg A. DNA replication. J. Biol. Chem., 263, 1-4, 1988.
LeBowitz J., McMacken R. The E. coli dnaB replication protein is a DNA helicase. J. Biol. Chem., 261, 4738-4748, 1986.
31. Modrich P. DNA mismatch correction. Annu. Rev. Biochem., 56, 435-466, 1987.
Radman M., Wagner R. The high fidelity of DNA duplication. Sci. Am., 259(2), 40-47, 1988.
32. Bramhill D., Kornberg A. Duplex opening by dnaA protein at novel sequences in initiation of replication at the origin of the E. coli chromosome. Cell, 52, 743-755, 1988.
Dodson M. et al. Specialized nucleoprotein structures at the origin of replication of bacteriophage lambda: localized unwinding of duplex DNA by a six-protein reaction. Proc. Natl. Acad. Sci. USA, 83, 7638-7642, 1986.
Zyskind J. W., Smith D. W. The bacterial origin of replication, ori C. Cell, 46, 489-490, 1986.
33. DiNardo S., Voelkel K., Sternglanz R. RNA topoisomerase II mutant of Saccharomyces cerevisiae: topoisomerase II is required for segregation of daughter molecules at the termination of DNA replication. Proc. Natl. Acad. Sci. USA, 81, 2616-2620, 1984.
Wang J.C. DNA topoisomerases. Annu. Rev. Biochem., 54, 665-698, 1985.
34. Campbell J. L. Eucaryotic DNA replication. Annu. Rev. Biochem., 55, 733-771, 1986.
35. Low К. В. ed. The Recombination of Genetic Material. San Diego, Academic Press, 1988.
Stahl F. W. Genetic recombination. Sci. Am., 256(2), 90-101, 1987.
Whitehouse H.L.K. Genetic Recombination: Understanding the Mechanisms. New York, Wiley, 1982.
36. Dressier D., Potter H. Molecular mechanisms of genetic recombination. Annu. Rev. Biochem., 51, 727-762, 1982.
Smith G. R. Mechanism and control of homologous recombination in Escherichia coli. Annu. Rev. Genet, 21, 179-201, 1987.
37. Wetmur J. G., Davidson N. Kinetics of renaturation of DNA. J. Mol. Biol., 31, 349-370, 1968.
38. Cox M. M., Lehman I. R. Enzymes of general recombination. Annu. Rev. Biochem., 56, 229-262, 1987.
Kowalczykowski S. C. Mechanistic aspects of the DNA strand exchange activity of E. coli recA protein. Trends Biochem. Sci., 12, 141-145, 1987.
Radding C. M. Recombination activities of E. coli recA protein. Cell, 25, 3-4, 1981.
39. Holliday R. A mechanism for gene conversion in fungi. Genet. Res., 5, 282-304, 1964.
Meselson M. S., Radding С. М. A general model for genetic recombination. Proc. Natl. Acad. Sci. USA, 72, 358-361, 1975.
Sigal N., Alberts B. Genetic recombination: the nature of a cross-strand exchange between two homologous DNA molecules. J. Mol. Biol., 71, 789-793, 1972.
40. Fincham J. R. S., Day P. R., Radford A. Fungal Genetics, 4th ed. Berkeley, University of California Press, 1979.
Kourilsky P. Molecular mechanisms of gene conversion in higher cells. Trends Genet, 2, 60-62, 1986.
41. Campbell A. Some general questions about movable elements and their implications. Cold Spring Harbor Symp. Quant. Biol., 45, 1-9, 1980.
Craig N. The mechanism of conservative site-specific recombination. Annu. Rev. Genet, 22, 77-105, 1988.
Grindley N. D., Reed R. R. Transpositional recombination in prokaryotes. Annu. Rev. Biochem., 54, 863-896, 1985.
Mizuuchi K., Craigie R. Mechanism of bacteriophage mu transposition. Annu. Rev. Genet, 20, 385-429, 1986.
42. Joklik W.K. ed. Virology, 2nd ed. Norwalk CT, Appleton and Lange, 1985. Shapiro JA. ed. Mobile Genetic Elements. New York, Academic Press, 1983.
Watson J. D., Hopkins N. H., Roberts J. W., Steitz J. A., Weiner A. M. Molecular Biology of the Gene, 4th ed. Menlo Park CA, Benjamin- Cummings, 1987. (Chapter 24 provides a modern overview of eucaryotic viruses.)
43. HersheyA.D., Chase M. Independent functions of viral protein and nucleic acid in growth of bacteriophage. J. Gen. Physiol., 36, 39-56, 1952.
Luria S. E., Darnell J. E., Baltimore D., Campbell A. General Virology, 3rd ed.
New York, Wiley, 1978. (The introductory chapter provides a historical overview of the development of virology.)
44. Simons K., Garoff H, Helenius A. How an animal virus gets in and out of its host cell. Sci. Am., 246(2), 58-66, 1982.
45. Gierer A., Schramm G. Infectivity of ribonucleic acid from tobacco mosaic virus. Nature, 177, 702-703, 1956.
46. Cohen S.S. Virus-Induced Enzymes. New York, Columbia University Press, 1968.
47. Kornberg A. DNA Replication. San Francisco, Freeman, 1980. (Chapters 14 and 15 provide up-to-date, beautifully illustrated description of the varieties of ways phage and animal viral DNA's replicate.)
48. Campbell A.M. Bacteriophage lambda as a model system. Bioessys, 5, 277-280, 1986.
Daniels D., Sanger F., Coulson A. R. Features of bacteriophage lambda: analysis of the complete nucleotide sequence. Cold Spring Harbor Symp. Quant. Biol., 47, 1009-1024, 1982.
Lwoff A. Lysogeny. Bacteriol. Rev., 17, 269-337, 1952.
49. Watson J.D., Hopkins N.H., Roberts J. W., Steitz J.A., Weiner A.M. Molecular Biology of the Gene, 4th ed. Menlo Park CA, Benjamin- Cummings, 1987. Chapter 26 describes the viruses that can cause tumors.)
50. Baltimore D. Virap DNA-dependent DNA polymerase. Nature, 226, 1209-1211, 1970.
GalloR.C. The AIDS virus. Sci. Am., 256(1), 46-56, 1987.
Temin H. M., Mizutani A. RNA-dependent DNA polymerase in virions of Rous sarcoma virus. Nature, 226, 1211-1213, 1970.
Varmus H. Retroviruses. Science, 240, 1427-1435, 1988.
51. Boeke J.D., Garfinkel D.J., Styles C.A., Fink G.R. Ту elements transpose through an RNA intermediate. Cell, 40, 491-500, 1985.
Fink G. R., Boeke J. D., Garfinkel D. J. The mechanisms and consequences of retrotransposition. Trends Genet., 2, 118-123, 1986.
52. Cohen S.N., Shapiro J.A. Transposable genetic elements. Sci. Am., 242(2), 40-49, 1980.
Mizuuchi K. Mechanism of transposition of bacteriophage Mu: polarity of the strand transfer reaction at the initiation of transposition. Cell, 39, 395-404, 1984.
53. Novick R.P. Plasmids. Sci. Am., 243(6), 102-127, 1980.
Reisner D., Gross H.J. Viroids. Annu. Rev, Biochem., 54, 531-564, 1985.
Rossmann M.G. The evolution of RNA viruses. Bioessays, 7, 99-103, 1987.
54. Drlica K. Understanding DNA and Gene Cloning. New York, Wiley, 1984.
Sambrook J., Fritsch E. F., Maniatis T. Molecular Cloning: A Laboratory Manual, 2nd ed. Cold Spring Harbor, NY, Cold Spring Harbor Laboratory, 1989.
Watson J.D., Tooze J., Kurtz D. T. Recombinant DNA: A Short Course. New York, W.H. Freeman, 1983.
55. Smith H. O. Nucleotide sequence specifity of restriction endonucleases. Science, 205, 455-462, 1979.
56. Cohen S., Chang A., Boyer H., Helling R. Construction of biologically functional bacterial plasmids in vitro. Proc. Natl. Acad. Sci. USA, 70, 3240-3244, 1973.
57. Maniatis T. et al. The isolation of structural genes from libraries of eucaryotic DNA. Cell, 15, 687-701, 1978.
Maniatis Т., Kee S. G., Efstratiadis A., Kafatos F. C. Amplification and characterization of a ß-globin gene synthesized in vitro. Cell, 8, 163182, 1976.
58. Hedrick S. M., Cohen D. I., Nielsen E. A., Davis M. M. Isolation of cDNA clones encoding T cell-specific membrane-associated proteins. Nature, 308, 149-153, 1984.
59. Grunstein M., Hogness D. S. Colony hybridization: a method for the isolation of cloned DNAs that contain a specific gene. Proc. Natl. Acad. Sci. USA, 72, 3961-3965, 1975.
Suggs S. V., Wallace R.B., Hirose Т., Kawashima E.H., Itakura K. Use of synthetic oligonucleotides as hybridization probes: isolation of cloned sequences for human ß2-microglobulin. Proc. Natl. Acad. Sci. USA, 78, 6613-6617, 1981.
60. Coulson A., Sulston J., Brenner S.. Karn J. Toward a physical map of the genome of the nematode Caenorhabaitis elegans. Proc. Natl. Acad. Sci. USA, 83, 7821-7825, 1986.
Proustka A., Lehrach H. Jumping libraries and linking libraries: the next generation of molecular tools in mammalian genetics. Trends Genet., 2, 174-179, 1986.
61. Pelham H.R.B., Jackson R. J. An efficient mRNA-dependent translation system from reticulocyte lysates. Eur. J. Biochem., 67, 247-256, 1976.
62. Geysen H. M. et al. Chemistry of antibody binding to a protein. Science, 235, 1184-1190, 1987.
Gilbert W., Villa-Komaroff L. Usefull proteins from recombinant bacteria. Sci. Am., 242(4), 74-94, 1980. Yokata T. et al. Use of a cDNA expression vector for isolation of mouse interleukin 2 cDNA clones: expression of T-cell growth-factor activity after transfection of monkey cells. Proc. Natl. Acad. Sci. USA, 82, 6872, 1985.
Young R. A., Davis R. W. Efficient isolation of genes by using antibody probes. Proc. Natl. Acad. Sci. USA, 80, 1194-1198, 1980.
63. Smith M. In vitro mutagenesis. Annu. Rev. Genet., 19, 423-462, 1985.
64. Hinnen A., Hicks J. В., Fink G. R. Transformation of yeast. Proc. Natl. Acad. Sci. USA, 75, 1929-1933, 1978.
Palmiter R. D., Brinster R. L. Germ line transformation of mice. Annu. Rev. Genet, 20, 465-499, 1986.
Spradling A.C., Rubin G. M. Transposition of cloned P elements into Drosophila germ line chromosomes. Science, 218, 341-347, 1982.
Thomas K. R., Capecchi M. R. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell, 51, 503-512, 1987.
65. Marx J.L. Multiplying genes leaps and bounds. Science, 240, 1408-1410, 1988.
Saiki R. K. et al. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science, 239, 487-491, 1988.
66. Botstein D., White R. L., Skolnick M., Davis R. W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet., 32, 314-331, 1980. Mapping and Sequencing the Human Genome. Washington DC, National Academy Press, 1988.
White R., LaLouel J.-M. Chromosome mapping with DNA markers. Sci. Am. 258(2), 40-49, 1988.