Основы молекулярной биологии. Часть 2: Молекулярные генетические механизмы - А.Н. Огурцов 2011
Геномика и протеомика
Кластерный анализ множественной экспрессии генов
Окончательные выводы о том, существуют или нет общие регуляторы для генов, которые демонстрируют синхронные изменения в экспрессии, а, следовательно, являются или нет эти гены функционально родственными, не могут быть сделаны только на основе эксперимента с микроматрицами ДНК.
Например, многие из обнаруженных отличий в экспрессии генов дрожжей при замене глюкозы на этанол, могут быть косвенным следствием многих различных изменений в физиологии клетки, которые происходят вследствие переноса клетки из одной среды в другую. Другими словами, изменение в экспрессии генов, которые демонстрируют синхронный отклик в эксперименте с микроматрицей ДНК, может быть вызваны совершенно различными причинами, а, следовательно, эти гены могут иметь совершенно различные биологические функции.
Решить эту проблему можно, комбинируя информацию серии нескольких различных экспрессионных экспериментов с тем, чтобы найти те гены, которые аналогично регулируются при различных воздействиях, причем синхронно во времени.
Например, одновременное исследование экспрессии 8600 генов в течении 24 часов после того как фибробласты человека были помещены в оптимальную для роста клеток сыворотку (а это более 10000 измерений кинетики флуоресценции отдельных ячеек матриц ДНК) позволило определить родственные связи разных генов, помогло (с помощью, конечно, компьютерной обработки) структурировать полученные данные и сгруппировать в "кластеры" гены, которые демонстрируют аналогичную экспрессию во времени (рисунок 120).
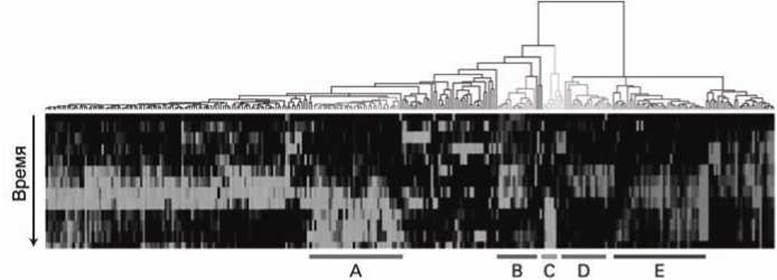
Рисунок 120 - Кластерный анализ данных множественных экспериментов по экспрессии генов методом микроматриц ДНК.
Примечательно, что такой кластерный анализ сгруппировал наборы генов, которые кодируют белки, участвующие в общеклеточных процессах таких, как биосинтез холестерола или клеточный цикл.
На рисунке 120 прописными английскими буквами обозначены такие кластеры генов, кодирующие белки, которые участвуют в клеточных процессах: А - биосинтез холестерола, В - клеточный цикл, С - немедленный отклик, D - сигнальные функции и ангиогенез (развитие кровеносной системы), Е - заживление ран и видоизменение тканей.
Поскольку гены, демонстрирующие идентичную или подобную картину регуляции, как правило, кодируют функционально родственные белки, кластерный анализ комбинированных данных серий экспрессионных микроматричных экспериментов является дополнительным методом для определения функций новых, недавно идентифицированных генов.
Такой подход позволяет комбинировать любое число различных экспериментов. При этом каждый новый эксперимент будет позволять заново проводить (уточнять) кластерный анализ, детализируя кластерную структуру генома.
В заключение отметим, что наглядным проявлением стремительного роста числа исследований в области геномики и протеомики в последнее время является появление новых специализированных научных журналов, посвященных сравнительному анализу геномов и белков. Так, например, с 2006 года издательство Elsevier начало выпуск журнала "Comparative Biochemistry and Physiology, Part D, Genomics and Proteomics", целиком посвященного такого рода исследованиям.
ВЫВОДЫ
Функции белка, который ещё не выделен в чистом виде, могут быть предсказаны на основе подобия его аминокислотной последовательности с аминокислотными последовательностями белков, чьи функции уже известны.
Компьютерный алгоритм BLAST проводит быстрый поиск в базе данных расшифрованных белковых последовательностей и находит участки подобия исследуемого (нового) белка с уже исследованными белками.
Белки с общими функциональными мотивами могут и не быть идентифицированными в стандартном BLAST-поиске. Для определения этих коротких участков белковой цепи используют базы данных белковых мотивов (повторов).
Белковые семейства объединяют белки которые ведут свою "родословную" от общего белка-предка. Гены, которые кодируют эти белки, и которые составляют соответствующее семейство генов, возникли от гена-предка вследствие его дупликации и последующей дивергенции в ходе видообразования.
Родственные гены и кодируемые ими белки, которые возникли вследствие дупликации генов называются паралогичными; те же, которые возникли в ходе видообразования называются ортологичными. Ортологичные белки, как правило, имеют подобные функции.
Открытые рамки считывания (ORF) представляют собой участки геномных ДНК, содержащие более 100 кодонов, которые расположены между старт и стоп кодонами.
Компьютерный поиск открытых рамок считывания в полных геномах бактерий и дрожжей правильно идентифицирует большинство генов, которые кодируют белки. Однако, вследствие более сложной организации структуры генов у человека и других высших эукариот, для идентификации вероятных генов в таких геномных последовательностях необходимо использовать дополнительные данные.
Проведенный полный анализ геномов нескольких разных организмов показал, что биологическая сложность не связана напрямую с числом белковых генов.
Анализ с помощью микроматриц ДНК одновременно детектирует относительный уровень экспрессии тысяч генов в разных типах клеток или в одной клетке, но при различных условиях.
Кластерный анализ результатов множественных экспериментов методом микроматриц ДНК может определить гены, регуляция которых подобна в различных условиях. Такие одинаковым образом регулируемые гены обычно кодируют белки, которые имеют биологически сходные (или связанные) функции.
Вопросы для самоконтроля
1. Какие генные последовательности называются гомологичными?
2. Какие генные последовательности называются паралогичными и в чем их отличие от ортологичных генных последовательностей?
3. Какие генные последовательности называются ортологичными и в чем их отличие от паралогичных генных последовательностей?
4. Что такое филогенетическая схема (кладограмма)?
5. Что называется открытой рамкой считывания?
6. Назовите основные причины того, что размер генома не коррелирует с биологической сложностью организма?
7. Как устроена микроматрица ДНК?
8. Что определяют методом кластерного анализа множественной экспрессии генов?