ИММУНОЛОГИЯ - Ройт А. - Мир 2000
Глава 16. Противовирусный иммунитет
ИММУНОПАТОЛОГИЯ
Иммунный ответ на вирусные антигены может вызывать повреждения тканей
Нарушения, связанные с иммунными комплексами. Иммунные комплексы могут появляться в различных жидкостях организма или на поверхности клеток, чаше всего при хронических, а также при персистентных инфекциях, вызванных, например, вирусами лимфоцитарного хориоменингита (LCMV. от англ. lymphocytic choriomeningitis virus) либо гепатита В. При избытке вирусного антигена антитела теряют способность нейтрализовывать вирусы; вместо этого они образуют иммунные комплексы, которые оседают в почках или в кровеносных сосудах других органов и вызывают там воспалительные реакции, чреватые повреждением тканей, например такие, как гломерулонефрит (см. гл. 25).
Связывание вирусов антителами, лишенными нейтрализующей активности, иногда имеет еще одно необычное патологическое следствие; эти иммунные комплексы в результате взаимодействия с Fc-рецептором поглощаются макрофагами, в которых инфекционность вируса усиливается. Это можно наблюдать при инфекции, вызванной вирусом денге. С Fc-рецепторными взаимодействиями иммунных комплексов, вызывающими гиперактивацию системы комплемента, связан также патогенез геморрагической лихорадки и шокового синдрома денге.
Повреждение тканей хозяина цитотоксическими Т-клетками. При любой вирусной инфекции некоторая часть тканевых повреждений вызвана Т-клеточной активностью. Иногда в эксперименте они настолько существенны, что могут вызвать гибель животного. Яркий пример этого — поражение клеток центральной нервной системы мыши цитотоксическими Т-клетками при иммунном ответе на заражение LCMV (рис. 16.11). Удаление Т-клеток спасает животных от гибели; таким образом, именно Т-клетки, а не вирусы, повреждают ткани мозга. Подобный механизм предположительно действует в патогенезе хронического активного гепатита у человека.
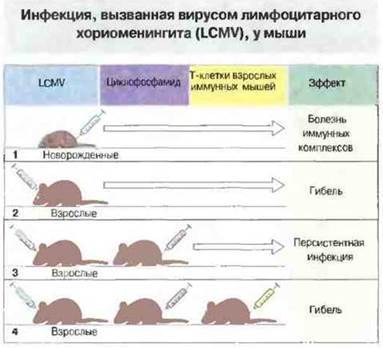
Рис. 16.11. При заражении мышей вирусом лимфоцитарного хориоменингита (LCMV) исход инфекции зависит от иммунного статуса животного. У новорожденных мышат (1) заражение приводит к хроническому выделению вирионов и болезни иммунных комплексов, которая проявляется как гломерулонефрит и васкулит. У взрослых животных (2) внутримозговое заражение вызывает гибель в результате активности собственных Т-клеток; на эту причину указывает то, что в случае подавления иммунитета циклофосфамидом (3) мыши выживают, хотя инфекция становится персистирующей. «Протективный» эффект циклофосфамида можно отменить переносом Т-клеток от иммунных животных (4).
Вирусы способны инфицировать клетки иммунной системы
Некоторые вирусы (например, HIV — от human immunodeficiency virus, или ВИЧ, вирус иммунодефицита человека) непосредственно инфицируют лимфоциты и макрофаги, вызывая патогенный эффект. Кроме того, иммунокомпетентные клетки служат для вирусов благоприятным местом персистенции. Вирусы п неинфекционной форме локализуются в покоящихся лейкоцитах, активация которых может вызвать и реактивацию вирусов с репликацией инфекционных вирионов. Примеры вирусов, заселяющих В-клетки, Т-клетки и макрофаги, приведены на рис. 16.12.

Рис. 16.12. Некоторые вирусы неопределенно долго порепетируют в иммунокомпетентных клетках. Периодически такая инфекция может приводить к патологическим последствиям, например к гибели клеток (ВИЧ) или их злокачественной трансформации (EBV, HTLV-1).
Вирус иммунодефицита человека инфицирует Т-клетки CD4+. В предыдущих разделах часто упоминался в качестве примера ВИЧ — ретровирус, вызывающий синдром приобретенного иммунодефицита (СПИД). Для этой инфекции характерны продолжительный бессимптомный период, неэффективность иммунитета, непрерывная антигенная изменчивость вируса, его стремление заселить лимфоциты и клетки миелоидного происхождения, а также невропатологическая симптоматика в развернутый период болезни (см. гл. 21).
Т-клетки и макрофаги поглощают ВИЧ вследствие того, что вирусный гликопротеин gpl20 связывается с маркером CD4 и с определенными рецепторами для хемокинов. CCR3 и CCR5. Подобным же образом ВИЧ проникает в любую другую аптигенпрезентирующую клетку. Противовирусные антитела могут способствовать этому промессу, если клетка обладает Fc-рецептором. По сути это альтернативный способ внедрения вируса в фагоцитарные клетки или механизм, усиливающий проникновение, в том случае когда CD4 присутствует в малом количестве.
Период отсутствия клинических симптомов при ВИЧ-инфекции варьирует у разных больных и может быть весьма длительным: примерно у половины инфицированных ВИЧ-инфекция не прогрессирует в СПИД в течение 10 лет. В этот латентный период инфекции возбудитель присутствует в организме в форме провируса, встроенного в геномную ДНК хозяина, и транскрипции вирусной ДНК не происходит. Активацию вируса и начало транскрипции могут вызвать многие факторы. Например, in vitro воздействие ФИО и ИЛ-6 на латентно инфицированные культуры Т-клеток приводит к повышенной продукции инфекционных вирионов. Этот феномен, вероятно, имеет место и in vivo, так как моноциты ВИЧ-инфицированных больных часто выделяют указанные цитокины в патологически высоком количестве. Возможно, существует цикл высвобождения ФИО и ИЛ-6, в определенной фазе которого происходит усиление транскрипции вирусных генов (рис. 16.13). Репликация вируса ведет к инфицированию все большего числа клеток и выделению все большего количества цитокинов, причем in vitro ее стимулируют не только указанные, но и другие цитокины и лимфокины, а также митогены и форболовые эфиры. Элиминации вируса не происходит по различным причинам, в том числе из-за его латентной персистенции, мутирования (вызывающего быстрый антигенный дрейф) и прогрессирующей иммунологической недостаточности.
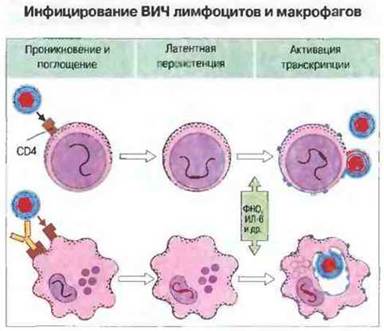
Рис. 16.13. Гликопротеин gр120, расположенный на поверхности ВИЧ, связывается с молекулой CD4 на плазматической мембране лимфоцита, активируя тем самым поглощение им вирусной частицы Таким же образом ВИЧ может проникать в макрофаги, экспрессирующие гораздо меньше молекул CD4. При этом поглощению его макрофагами способствуют противовирусные антитела, связывающиеся с фагоцитарными клетками через рецепторы для Fc. Вирус встраивается в геномную ДНК клеток хозяина и остается латентным до тех пор, пока определенные стимулы (например, цитокины) не активируют транскрипцию вирусных генов Но вообразованные вирусные частицы после сборки выходят из Т-клеток путем отпочковывания от цитоплазматической мембраны либо тем же способом отпочковывания проникают во внутриклеточные вакуоли макрофагов. Таким образом в макрофагах может накапливаться большое количество потенциально инфекционных вирусных частиц.
Вирусная инфекция может провоцировать аутоиммунные заболевания
Вирусный патогенез аутоиммунных болезней имеет несколько механизмов.
Индуцированное вирусами повреждение тканей. Некоторые вирусные инфекции вызывают повреждение тканей и последующую воспалительную реакцию, в результате которой начинают экспонироваться ранее «скрытые» собственные антигены; они могут пройти процессинг и быть презентированы клеткам иммунной системы. Это наблюдается, например, при инфекциях нервной системы, вызванных вирусом Тендера (пикорна-вирус мыши) и вирусом гепатита мыши, когда мишенями для антител и Т-клеток становятся компоненты миелиновой оболочки аксонов.
Молекулярная мимикрия. Иммунная система распознает как «чужое» аминокислотную последовательность вирусного белка, который гомологичен одному из белков организма-хозяина. В результате происходит срыв иммунологической толерантности к собственным скрытым антигенам и последующая атака иммунной системы против тканей хозяина (см. гл. 28). Модель такого патогенетического механизма аутоиммунопатологии может быть создана экспериментально, но доказательств его действия при естественной вирус-
Вопросы для размышления
■ За счет каких свойств вирусы способны ускользать от действия защитных механизмов организма-хозяина?
■ В клинику поступил маленький мальчик, страдающий диссеминированной герпесвирусной инфекцией. Какую иммунотерапию вы ему назначите и почему?
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
Borden E.G., Rosenzweig I.В., Byrne G.l. 1987. Interferons: from vires inhibitor to modulator of amino acid and lipid metabolism. Interferon Res. 7: 591.
Chisari F.V., Ferrari C. 1995. Hepatitis В virus immuno-pathogenesis. Annu. Rev. Immunol. 13: 29-60.
Clements J.E., Gidovin S.L., Montelaro R.C. et al. 1988. Antigenic variation in lentiviral disease. Annu. Rev. Immunol. 6: 139-159.
Doherty P C., Allan W., Eichelberg M. etal. 1992. Roles of а/p and y/8 T cell subsets in viral immunity. Annu. Rev. 10: 123-151.
Doherty P.C. 1993. Cell-mediated cytotoxicy. Cell. 75: 607.
Gooding L.R. 1992. Virus proteins that counteract host immune defences. Cell 71: 5-7.
Levy J.A. 1993. Pathogenesis of human immunodeficiency virus infection. Microbiol. Rev. 57: 183-289.
Mims C.A. 1986. Interactions of viruses with the immune system. Clin. Exp. Immunol. 66: 1-16.
Nash A.A., Cambouropoulos P. 1993. The immune response to herpes simplex virus. Semin. Virol. 4: 181-186.
Oldstone M.B.A. 1987. Molecular mimicry and autoimmune disease. Cell 50: 819-820.
Ramsay A.J. 1993. A case for cytokines as effector molecules in the resolution on virus infection. Immunol. Today 14: 155.
Sissons J.G., Oldstone M.B.A. 1980. Antibody-mediated destruction of virus-infected cells. Adv. Immunol. 31: 1.
Smith G.A. 1994. Virus strategies for evasion of the host response to infection. Trends. Microbiol. 2: 81-88.