Биологическая химия - Березов Т. Т., Коровкин Б. Ф. 1998
Обмен простых белков
Промежуточный обмен аминокислот в тканях
Трансаминирование аминокислот
Под трансаминированием подразумевают реакции межмолекулярного переноса аминогруппы (NH2—) от аминокислоты на а-кетокислоту без промежуточного образования аммиака. Впервые реакции трансаминирования (прежнее название «переаминирование») были открыты в 1937 г. советскими учеными А.Е. Браунштейном и М.Г. Крицман при изучении дезаминирования глутаминовой кислоты в мышечной ткани. Было замечено, что при добавлении к гомогенату мышц глутаминовой и пировиноградной кислот образуются а-кетоглутаровая кислота и аланин без промежуточного свободного аммиака; добавление аланина и а-кетоглутаровой кислоты приводило к образованию соответственно пировиноградной и глутаминовой кислот.
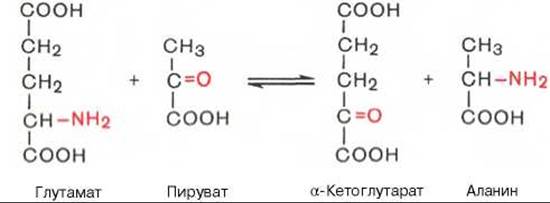
Реакции трансаминирования являются обратимыми и, как выяснилось позже, универсальными для всех живых организмов. Эти реакции протекают при участии специфических ферментов, названных А.Е. Браунштейном аминоферазами (по современной классификации, аминотрансферазы, или трансаминазы). Теоретически реакции трансаминирования возможны между любой амино- и кетокислотой, однако наиболее интенсивно они протекают в том случае, когда один из партнеров представлен дикарбоновой амино- или кетокислотой. В тканях животных и у микроорганизмов доказано существование реакций трансаминирования между монокарбоновыми амино- и кетокислотами. Донорами NН2-группы могут также служить не только а-, но и ß-, у- и ю-аминогруппы ряда аминокислот. В лаборатории А. Майстера доказано, кроме того, трансами- нирование глутамина и аспарагина с кетокислотами в тканях животных.
В переносе аминогруппы активное участие принимает кофермент трансаминаз пиридоксальфосфат (производное витамина В6; см. главу 5), который в процессе реакции обратимо превращается в пиридоксаминфосфат.
Механизм реакции трансаминирования. Общую теорию механизма ферментативного трансаминирования разработали советские ученые А.Е. Браунштейн и М.М. Шемякин. Одновременно подобный механизм был предложен американскими биохимиками Э. Снеллом и Д. Метцлером. Все трансаминазы (как и декарбоксилазы аминокислот) содержат один и тот же кофермент — пиридоксальфосфат. Для реакций трансаминирования характерен общий механизм. Специфичность трансаминаз обеспечивается белковым компонентом. Ферменты трансаминирования катализируют перенос NH2-группы не на а-кетокислоту, а сначала на кофермент пиридоксальфосфат. Образовавшееся промежуточное соединение (шиффово основание) подвергается внутримолекулярным превращениям (лабилизация а-водородного атома, перераспределение энергии связи), приводящим к освобождению а-кетокислоты и пиридоксаминфосфата; последний на второй стадии реакции реагирует с любой другой а-кетокислотой, что через те же стадии образования промежуточных соединений (идущих в обратном направлении) приводит к синтезу новой аминокислоты и освобождению пиридоксальфосфата. Опуская промежуточные стадии образования шиффовых оснований, обе стадии реакции трансаминирования можно представить в виде общей схемы:
![]()
Более подробно механизм действия трансаминаз представлен на рис. 12.3.
В связи с тем что во всех пиридоксалевых ферментах (включая трансаминазы) карбонильная группа кофермента (—СНО) оказалась связанной с ε-аминогруппой лизина белковой части, в классический механизм реакции трансаминирования А.Е. Браунштейн и Э. Снелл внесли следующее дополнение. Оказалось, что взаимодействие между субстратом, т.е. L-аминокислотой (на рисунке — аспартат), и пиридоксальфосфатом происходит не путем конденсации с выделением молекулы воды, а путем реакции замещения, при которой NH2-группа субстрата вытесняет ε-NН2-группу лизина в молекуле ферментного белка, что приводит к формированию пиридоксальфосфатного комплекса.
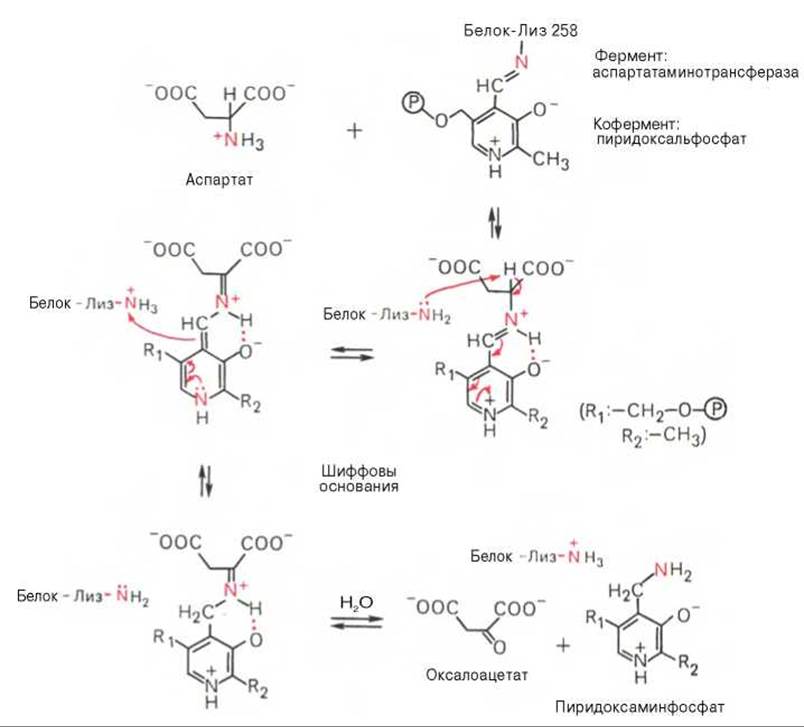
Рис. 12.3. Механизм действия пиридоксальфосфата в аспартатаминотрансферазе.
Существование представленного механизма реакции трансаминирования доказано разнообразными методами, включая методы спектрального анализа по идентификации промежуточных альдиминных и кетиминных производных пиридоксальфосфата.
Роль трансаминаз и реакций трансаминирования в обмене аминокислот.
Чрезвычайно широкое распространение трансаминаз в животных тканях, у микроорганизмов и растений, их высокая резистентность к физическим, химическим и биологическим воздействиям, абсолютная стереохимическая специфичность по отношению к L-аминокислотам, а также высокая каталитическая активность в процессах трансаминирования послужили предметом детального исследования роли этих ферментов в обмене аминокислот. Ранее было указано, что при физиологических значениях рН среды активность оксидазы L-аминокислот резко снижена. Учитывая это обстоятельство, а также высокую скорость протекания реакции трансаминирования, А.Е. Браунштейн выдвинул гипотезу о возможности существования в животных тканях непрямого пути дезаминирования аминокислот через реакции трансаминирования, названного им трансдезаминированием. Основой для выдвижения этой гипотезы послужили также данные Г. Эйлера о том, что в животных тканях из всех природных аминокислот с высокой скоростью дезаминируется только L-глутаминовая кислота в реакции, катализируемой высокоактивной и специфической глутаматдегидрогеназой.
Согласно гипотезе, получившей экспериментальное подтверждение, все или почти все природные аминокислоты (исключение составляет метионин) сначала реагируют с а-кетоглутаровой кислотой в реакции трансаминирования с образованием глутаминовой кислоты и соответствующей кетокислоты. Образовавшаяся глутаминовая кислота затем подвергается непосредственному окислительному дезаминированию под действием глутаматдегидрогеназы. Схематически механизм трансдезаминирования можно представить в следующем виде:
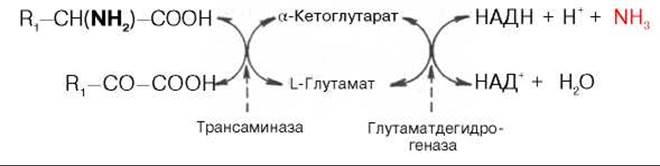
Суммарная реакция при этом следующая:
R1—CH(NH2)—COOH + НАД+ + H2O -> R—СО—СООН + НАДН2 + NH3.
Поскольку обе реакции (трансаминирование и дезаминирование глутаминовой кислоты) являются обратимыми, создаются условия для синтеза по существу любой аминокислоты, если в организме имеются соответствующие а-кетокислоты. Известно, что организм животных и человека не наделен способностью синтеза углеродных скелетов (а-кетокислот), так называемых незаменимых аминокислот; этой способностью обладают только растения и многие микроорганизмы.
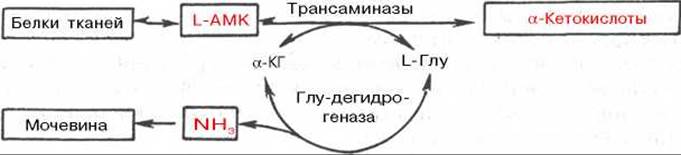
Рис. 12.4. Центральная роль трансаминаз L-аминокислот и глутаматдегидрогеназы в биосинтезе и распаде аминокислот в тканях животных.
АМК - аминокислоты; а-КГ - а-кетоглутарат.
Механизм, при помощи которого в живых организмах осуществляется синтез природных аминокислот из а-кетокислот и аммиака, был назван А.Е. Браунштейном трансреаминированием. Сущность его сводится к восстановительному аминированию а-кетоглутаровой кислоты с образованием глутаминовой кислоты (реакцию катализирует НАДФ-зависимая глутаматдегидрогеназа, работающая в режиме синтеза) и к последующему трансаминированию глутамата с любой а-кетокислотой. В результате образуется L-аминокислота, соответствующая исходной кетокислоте, и вновь освобождается а-кетоглутаровая кислота, которая может акцептировать новую молекулу аммиака. Роль реакций трансаминирования как в дезаминировании, так и в биосинтезе аминокислот может быть представлена в виде схемы:
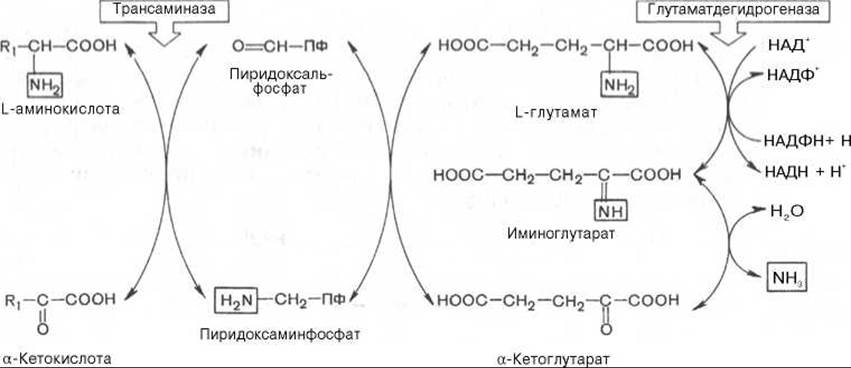
Таким образом, трансаминазы катализируют опосредованное через глутаматдегидрогеназу дезаминирование природных аминокислот (черные стрелки) и биосинтез аминокислот (красные стрелки). В более упрощенной форме роль этих ключевых ферментов азотистого обмена представлена на рис. 12.4.
Получены доказательства существования в организме теплокровных животных еще одного механизма непрямого (опосредованного) дезаминирования L-аминокислот, при котором Глу, Асп и АМФ выполняют роль системы переноса NН2-группы; гидролитическое дезаминирование АМФ приводит к образованию инозинмонофосфата (ИМФ) и аммиака:
![]()
Возможно, что в аналогичной системе в качестве промежуточного переносчика NH2-группы вместо АМФ участвует НАД.
Клиническое значение определения активности трансаминаз. Широкое распространение и высокая активность трансаминаз в органах и тканях человека, а также сравнительно низкие величины активности этих ферментов в крови послужили основанием для определения уровня ряда трансаминаз в сыворотке крови человека при органических и функциональных поражениях разных органов. Для клинических целей наибольшее значение имеют две трансаминазы — аспартат-аминотрансфераза (AcAT) и аланин-аминотрансфераза (АлАТ), катализирующие соответственно следующие обратимые реакции:
![]()
В сыворотке крови здоровых людей активность этих трансаминаз в тысячи раз ниже, чем в паренхиматозных органах. Поэтому органические поражения при острых и хронических заболеваниях, сопровождающиеся деструкцией клеток, приводят к выходу трансаминаз из очага поражения в кровь. Так, уже через 3—5 ч после развития инфаркта миокарда уровень АсАТ в сыворотке крови резко повышается (в 20—30 раз). Максимум активности обеих трансаминаз крови приходится на конец первых суток, а уже через 2—3 дня при благоприятном исходе болезни уровень сывороточных трансаминаз возвращается к норме. Напротив, при затяжном процессе или наступлении повторного инфаркта миокарда наблюдается новый пик повышения активности этих ферментов в крови. Этим объясняется тот факт, что в клинике трансаминазный тест используется не только для постановки диагноза, но и для прогноза и проверки эффективности лечения*. При поражениях клеток печени, например при гепатитах, также наблюдается гипертрансаминаземия (за счет преимущественного повышения уровня АлАТ), но она имеет более умеренный и затяжной характер, а повышение активности трансаминазы в сыворотке крови происходит медленно. При различного рода коронарной недостаточности (стенокардия, пороки сердца и др., кроме инфаркта миокарда) гипертрансаминаземия или не наблюдается, или незначительна. Определение активности трансаминаз в сыворотке крови при заболеваниях сердца следует отнести к дифференциально-диагностическим лабораторным тестам. Повышение уровня трансаминаз в сыворотке крови отмечено, кроме того, при некоторых заболеваниях мышц, в частности при обширных травмах, гангрене конечностей и прогрессивной мышечной дистрофии.
* В настоящее время с диагностической целью в клинике внутренних болезней широко используют наборы химических реактивов для быстрого (экспрессного) определения активности трансаминаз в сыворотке крови.
Превращения а-кетокислот. Образовавшиеся в процессе дезаминирования и трансдезаминирования а-кетокислоты подвергаются в тканях животных различным превращениям и могут вновь трансаминироваться с образованием соответствующей аминокислоты. Это так называемый синтетический путь превращения. Опыты с перфузией растворов а-кетокислот и аммиака через изолированную печень показали, что в оттекающей из печени жидкости действительно имеются соответствующие исходным кетокислотам L-аминокислоты. Открыты, кроме того, гликогенные, кетогенные и окислительные пути, ведущие к образованию соответственно глюкозы, жирных кислот, кетоновых тел и компонентов цикла трикарбоновых кислот (ЦТК). Эти процессы можно представить в виде общей сводной схемы:
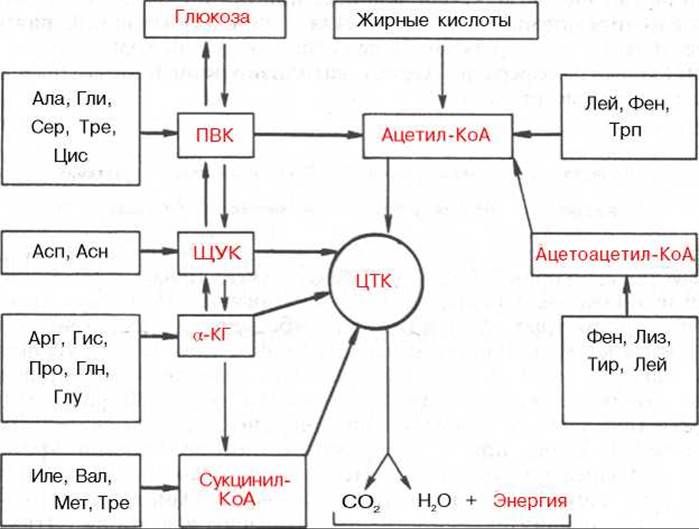
Углеродные скелеты аминокислот могут включаться в ЦТК через ацетил-КоА, пируват, оксалоацетат, а-кетоглутарат и сукцинил-КоА. Пять аминокислот (Фен, Лиз, Лей, Трп, Тир) считаются «кетогенными», поскольку они являются предшественниками кетоновых тел, в частности ацетоуксусной кислоты, в то время как большинство других аминокислот, обозначаемых как «гликогенные», служат в организме источником углеводов, в частности глюкозы. Подобный синтез углеводов de novo усиливается при некоторых патологических состояниях, например при сахарном диабете, а также при гиперфункции коркового вещества надпочечников и введении глюкокортикоидов (см. главу 8). Разделение аминокислот на «кетогенные» и «гликогенные» носит, однако, условный характер, поскольку отдельные участки углеродных атомов Лиз, Трп, Фен и Тир могут включаться и в молекулы предшественников глюкозы, например Фен и Тир — в фумарат. Истинно «кетогенной» аминокислотой является только лейцин.