Підручник - БІОЛОГІЧНА ХІМІЯ - Губський Ю.І. - 2000
Розділ II. ЗАГАЛЬНІ ЗАКОНОМІРНОСТІ МЕТАБОЛІЗМУ
ГЛАВА 7. ФЕРМЕНТИ II. МЕХАНІЗМИ КАТАЛІЗУ. КІНЕТИКА. РЕГУЛЯЦІЯ
7.2. КІНЕТИКА ФЕРМЕНТАТИВНИХ РЕАКЦІЙ. ІНГІБІТОРИ ФЕРМЕНТІВ
Ферментативна кінетика є розділом хімічної кінетики, що займається вивченням впливу різних хімічних та фізико-хімічних факторів на швидкість реакцій.
Вона вивчає, зокрема, залежність швидкостей ферментативних реакцій від концентрацій ферменту, субстрату, рН та температури середовища, дії активаторів та інгібіторів.
Загальне рівняння односубстратної ферментативної реакції:
![]()
З урахуванням взаємодії ферменту із субстратом у ході каталітичного акту (теорія Міхаеліса-Ментен) рівняння ферментативного перетворення субстрату набуває вигляду:
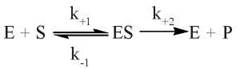
Для характеристики утворення фермент-субстратного комплексу використовується субстратна константа, або константа дисоціації комплексу Міхаеліса:
![]()
Відношення між сумою констант швидкостей реакцій зворотного розпаду (k-1) і розщеплення комплексу з утворенням продуктів реакції (k+2) та константою швидкості утворення фермент-субстратного комплексу (k+1) називається константою Міхаеліса (Кm):
![]()
Кm має розмірність концентрації (моль/л) і кількісно визначає спорідненість ферменту із субстратом — чим активніший фермент, тим нижче значення його Кm. Значення К для різних ферментів коливаються в широкому діапазоні — від 10-6 моль/л для високоактивних ферментів (наприклад, пероксидази) до 10-2 для малоактивних протеаз.
Залежність швидкості реакції від концентрації ферменту та субстрату
Концентрації ферменту та субстрату є величинами, що найбільш часто змінюються в умовах будь-якої біохімічної ферментативної реакції.
Цілком зрозуміло, що швидкість ферментативної реакції буде прямо пропорційно залежати від концентрації ферменту, а саме:
V = k · [E] ,
тобто збільшення в клітині рівня певного ферментного білка повинно супроводжуватися зростанням швидкості реакції, що каталізується цим ферментом.
Більш складною є залежність швидкості ферментативної реакції від концентрації субстрату. Графічно ця залежність зображується гіперболою, що подана на рис. 7.5. Як видно з ходу гіперболи, ця залежність має складний характер: при низьких концентраціях субстрату швидкість реакції прямо пропорційна його концентрації (реакція 1-го порядку), а при високих концентраціях досягається ефект насичення, тобто незалежність V від [S].
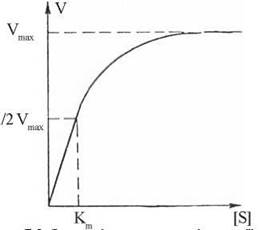
Рис. 7.5. Залежність швидкості реакції від концентрації субстрату (гіпербола Міхаеліса-Ментен).
Рівняння залежності V від [S], або рівняння Міхаеліса-Ментен таке:
![]()
У випадку, коли V = 1/2 Vmax, маємо:
![]()
звідси:
![]()
та після відповідних перетворень:
Km = [S].
Константа Кm дорівнює концентрації субстрату, при якій швидкість реакції становить половину від максимальної (рис. 7.5).
Рівняння Міхаеліса-Ментен можна отримати на основі аналізу кінетики реакції взаємодії ферменту із субстратом — за Брігсом та Холдейном.
Розглядаємо вихідне кінетичне рівняння.
Введемо такі позначення:
![]()
[Е] — загальна кількість ферменту (у вільній та зв’язаній формах);
[ЕS] — концентрація ферменту, що входить до складу фермент-субстратного комплексу;
([Е] - [ЕS]) — концентрація вільного, не зв’язаного з субстратом, ферменту.
Вважаємо також, що [S] » [Е].
Швидкість реакції утворення фермент-субстратного комплексу:
![]()
Швидкість реакції розщеплення фермент-субстратного комплексу:
![]()
Після встановлення в системі стаціонарного стану:
![]()
Враховуючи (2) та (3), можна вважати, що:
![]()
Розв’язуємо рівняння (4) через ![]()
![]()
З рівняння (5) знаходимо концентрацію [ES] в стаціонарному стані:
![]()
Загальну швидкість ферментативної реакції, тобто швидкість утворення продукту, можна подати як:
![]()
В умовах, коли
![]()
(тобто весь фермент входить до складу фермент-субстратного комплексу), швидкість реакції V = Vmax, тобто, враховуючи (7, 8):
![]()
Підставляючи значення [Е] та [ES] з (7) та (9) до рівняння (6), легко показати, що:
![]()
Обробка рівняння Міхаеліса-Ментен за методом подвійних зворотних величин дає змогу представити залежність V від [S] прямою лінією — рівняння Лайнуівера-Берка:
![]()
Перевагою цього рівняння є прямо пропорційна залежність між 1/V та 1/[S] (рис.7.6), яка дозволяє легко отримати значення кінетичних констант Кm та Vmax, що неможливо при аналізі звичайної гіперболи Міхаеліса-Ментен.
Із рівняння Лайнуївера-Берка та графіка (рис. 7.6) очевидно, що кутовий коефіцієнт прямої (тангенс кута нахилу) дорівнює Km/Vmax. Значення цих констант легко знаходять на графіку.
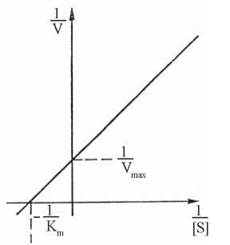
Рис. 7.6. Залежність 1/v від 1/[S] — графік Лайнуівера-Берка.
Залежність швидкості реакції від рН та температури
Вплив рН на активність ферментів
Кожен фермент має свій рН-оптимум, тобто значення рН середовища, при якому його каталітична активність максимальна. «Дзвоноподібна» залежність активностей ферментів від змін рН визначається їх білковою природою, зсувами в дисоціації іоногенних груп та (при екстремальних значеннях рН) розвитком конформаційних змін молекул.
Більшість внутрішньоклітинних та тканинних ферментів організму людини найактивніші в нейтральному, слаболужному або слабокислому середовищі (звичайно, в межах рН між 5,0 та 9,0). Ферментами з оптимумами при екстремальних значеннях рН є пепсин (рНопт = 1-2) і аргіназа (рНопт = 10-11).
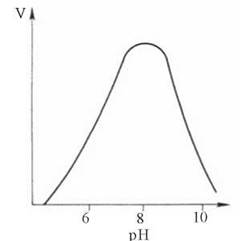
Рис. 7.7. Типова рН-залежність швидкості ферментативної реакції.
Вплив температури на активність ферментів
Ферменти, відповідно до своєї білкової природи, є термочутливими та термолабільними утвореннями:
- зростання температури до оптимальних значень (для більшості ферментів — у межах 37-40 °C) cупроводжується збільшенням швидкості ферментативної реакції відповідно за рівнянням Арреніуса (за рахунок частіших ефективних зіткнень між молекулами); ступінь збільшення швидкості реакції при зростанні t° на 10 °C позначають, як температурний коефіцієнтQ10;
- при збільшенні температури вище оптимального значення швидкість ферментативної реакції різко зменшується за рахунок конформаційних (денатураційних) змін у структурі ферментного білка.
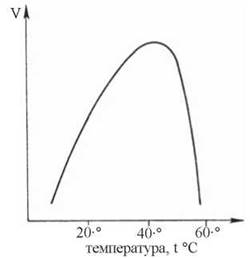
Рис. 7.8. Вплив температури на швидкість ферментативної реакції.
Інгібітори ферментів
Інгібітори — хімічні сполуки, що зменшують каталітичну активність ферментів. На відміну від речовин, які інактивують ферменти за рахунок їх денатурації (концентровані кислоти та луги, солі важких металів у високих концентраціях), дія інгібіторів є специфічною стосовно певних ферментів або груп ферментів, вони мають низьку концентрацію.
Залежно від характеру змін, що відбуваються в молекулі ферменту, розрізняють:
- зворотне інгібірування, що описується таким рівнянням взаємодії ферменту з інгібітором I:
![]()
- незворотне інгібірування:
![]()
Зворотне інгібірування ферментів, залежно від механізму взаємодії ферменту з інгібітором, поділяється на конкурентне та неконкурентне.
Конкурентне інгібірування.
Конкурентне інгібірування спричиняють ліганди, що за своєю хімічною структурою близькі до субстрату і взаємодіють із тим самим активним центром на молекулі ферменту, що і субстрат, утворюючи комплекс EI:
![]()
Класичним прикладом конкурентного інгібітора є малонова кислота НООС- СН2-СООН, яка протидіє зв’язуванню активним центром ферменту сукцинат-дегідрогенази справжнього субстрату — янтарної кислоти (сукцинату) НООС-СН2-СН2-СООН. Конкурентне інгібірування викликають різні антиметаболіти, тобто сполуки, близькі за будовою до справжніх клітинних метаболітів: антивітаміни; речовини, близькі до амінокислот, пуринових та піримідинових основ і нуклеотидів. У зв’язку з високою біологічною активністю деякі антиметаболіти застосовують як антибактеріальні засоби (сульфаніламіди, антибіотики), протипухлинні препарати.
Конкурентне інгібірування ферменту можна перебороти за рахунок підвищення концентрації субстрату в інкубаційному середовищі. Кінетичний аналіз за Лайнуівером-Берком свідчить, що конкурентні інгібітори збільшують константу Міхаеліса Кm ферменту і не впливають на Vmax реакції (рис. 7.9).
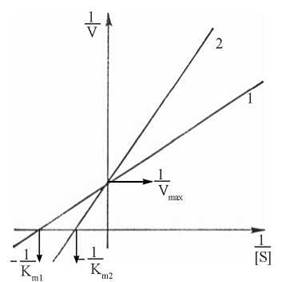
Рис. 7.9. Конкурентне інгібірування ферменту: 1 — без інгібітора; 2 — в присутності інгібітора.
Неконкурентне інгібірування.
Неконкурентні інгібітори не мають структурної подібності до субстрату. Вони реагують з іншими, відмінними від активних центрів, ділянками на молекулі ферменту і можуть зв’язуватися не тільки з вільним ферментом, а й із фермент-субстратним комплексом:
![]()
Приєднання неконкурентного інгібітора до ферменту зменшує його активність (максимальну швидкість реакції (Vmax), але не впливає на спорідненість ферменту із субстратом (Кm) (рис. 7.10).
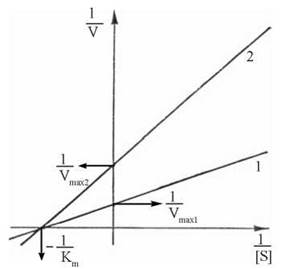
Рис. 7.10. Неконкурентне інгібірування ферменту: 1 — без інгібітора; 2 — в присутності інгібітора.
Неконкурентними інгібіторами є іони важких металів (Cu2+, Hg2+, Ag+) та їх похідні, що зворотно зв’язуються із SH-групами цистеїну в молекулах ферментів:
![]()
Незворотне інгібірування ферментів — процес, що відбувається внаслідок руйнування або незворотної хімічної модифікації однієї чи декількох функціональних груп ферменту. Незворотні інгібітори мають властивості клітинних отрут.
Прикладом такої модифікації молекули ферменту є дія алкілуючих агентів (зокрема, йодацетаміду), що незворотно реагують із каталітично активними SH-групами:
![]()
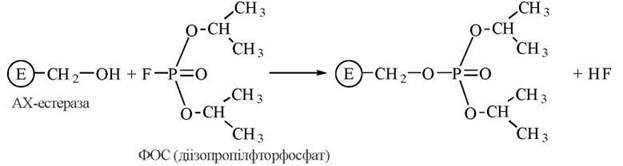
Практично важливим прикладом незворотного інгібірування ферменту шляхом ковалентного зв’язування інгібітора з активним центром є вплив фосфорорганічних сполук (ФОС) на активність ферменту ацетилхолінестерази (АХ-естерази). Препарати ФОС є високотоксичними отрутами відносно комах (пестициди) та теплокровних тварин, механізм антихолінестеразного ефекту яких полягає у взаємодії з ОН-групою серину в активному центрі ферменту: