Підручник - БІОЛОГІЧНА ХІМІЯ - Губський Ю.І. - 2000
Розділ III. МЕТАБОЛІЗМ ОСНОВНИХ КЛАСІВ БІОМОЛЕКУЛ
ГЛАВА 18. МЕТАБОЛІЗМ АМІНОКИСЛОТ. ІІ. СПЕЦІАЛІЗОВАНІ ШЛЯХИ ОБМІНУ
18.4. МЕТАБОЛІЗМ ПОРФІРИНІВ
Порфірини та їх комплекси з металами — металопорфірини — є простетичними групами багатьох гемопротеїнів — білків, які беруть участь в окислювально-відновлювальних реакціях у тваринних та рослинних клітинах. Представниками гемопротеїнів, що містять металопорфіринові групи, є Fe2+-вмісні гемоглобін (О2 — транспортуючий білок еритроцитів) і міоглобін (О2 — запасаючий білок м’язів), (Fe2+-Fe3+)- та (Cu1+-Cu2+)-вмісні цитохроми, (Fе2+-Fе3+)-вмісні ферменти каталаза, пероксидази, триптофанпіролаза, Mg2+-вмісний хлорофіл рослин.
Структура порфіринів
Порфірини — сполуки циклічної будови, основою структури яких є ароматична гетероциклічна система — порфін. Порфін, в свою чергу, є тетрапіролом, який утворюється при сполученні між собою метенільними (-СН=) містками чотирьох кілець азотистого гетероциклу піролу.
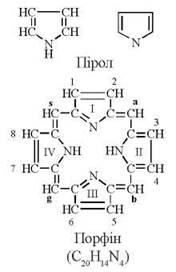
Будова піролу та порфіну.
Порфірини — похідні порфіну, заміщені в положеннях 1,2,3,4,5,6,7,8.
Природні порфірини, що в комплексі з металами входять до складу багатьох фізіологічно важливих гемопротеїнів, — це сполуки, в яких вісім атомів водню порфіринового ядра заміщені різними вуглеводневими радикалами. Залежно від будови бічних радикалів, розрізняють декілька класів порфіринів: уропорфірини, копропорфірини, протопорфірини, етіопорфірини, гематопорфірини, мезопорфірини, дейтеропорфірини. Кожен клас порфіринів містить декілька ізомерів, що позначаються літерами латинського алфавіту.
Метаболічні попередники порфіринів мають назву порфіриногенів (уропорфіриногени, копропорфіриногени тощо). На відміну від порфіринів, порфіриногени не містять спряжених метенільних структур (в їх молекулах піроли сполучені насиченими метиленовими (-СН2-) містками); вони є безбарвними сполуками і перетворюються на забарвлені порфірини при ферментативному або неферментативному (під дією кисню повітря) окисленні.
До складу гему кисеньзв’язуючих білків організму людини — гемоглобіну, міоглобіну та цитохромів дихальних ланцюгів входить порфірин, що позначається як протопорфірин ІІІ (за старою номенклатурою Фішера цей порфірин класифікувався як протопорфірин ІХ — номенклатурне позначення, що використовується і в даний час).
Синтез порфіринів
Біосинтез порфіринів тісно пов’язаний з амінокислотним метаболізмом: попередниками в утворенні піролових кілець порфіринів є гліцин та сукциніл-КоА. Послідовність реакцій синтезу така:
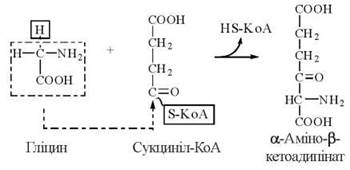
1. Взаємодія гліцину з сукцинілом-КоА з утворенням α-аміно-β-кетоадипінової кислоти:
2. Декарбоксилювання α-аміно-β-кетоадипінової кислоти з утворенням δ-амінолевулінової кислоти (ДАЛК):
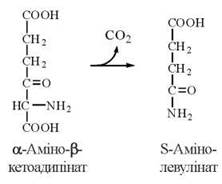
Обидві зазначені реакції, що призводять до утворення ДАЛК, каталізуються ферментом δ-амінолевулінатсинтазою (ДАЛК-синтазою). ДАЛК-синтаза є ПАЛФ-вмісним ферментом, що локалізований у мітохондріях та ендоплазматичному ретикулумі, в найбільшій кількості — в клітинах печінки (де він бере участь в синтезі простетичних груп мітохондріальних цитохромів та мікросомального цитохрому Р-45О), кісткового мозку та незрілих еритроцитах (ретикулоцитах).
3. Взаємодія двох молекул δ-амінолевулінату в реакції дегідратації з утворенням циклічної структури — порфобіліногену — безпосереднього метаболічного попередника порфіринів:
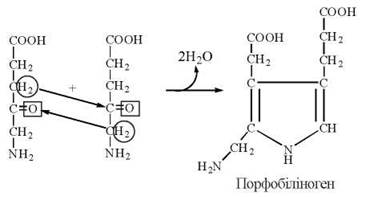
δ-Амінолевулінат, втрачаючи аміногрупу, може також перетворюватися на субстрати цитратного циклу — α-кетоглутарат, а потім — сукциніл-КоА, що дозволяє представити процес синтезу порфобіліногену у вигляді метаболічного циклу — цикл Шеміна-Рітенберга(D. Shemin, D. Rittenbeig, 1944):
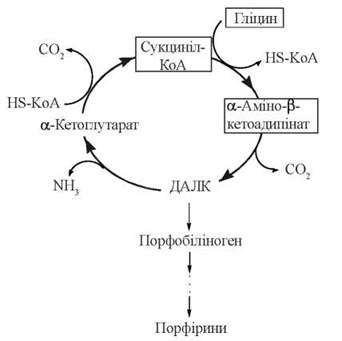
4. Синтез тетрапірольних структур.
Конденсація чотирьох порфобіліногенових одиниць призводить до утворення різних типів порфіринів. У разі спрямованості процесу на синтез гему — простетичної групи гемоглобіну та деяких цитохромів, генерація порфіринового циклу гему — протопорфірину IX відбувається в результаті такої послідовності реакцій:
4.1. Синтез із чотирьох молекул порфобіліногену уропорфіриногену III:
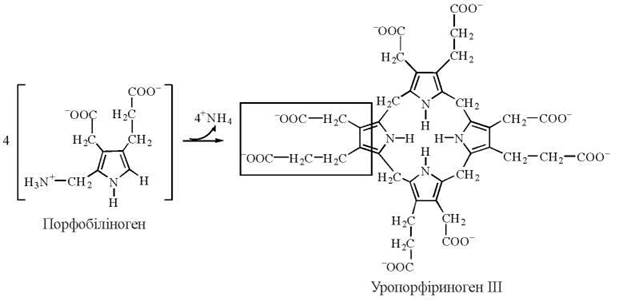
Реакція відбувається за участю двох білків:
- ферменту уропорфіриноген-синтази (порфобіліногендезамінази);
- білка уропорфіриноген ІІІ-косинтази.
Каталітична дія самого по собі ферменту уропорфіриноген-синтази, тобто у відсутності косинтази, призводить до утворення нефізіологічного ізомерного порфірину — уропорфіриногену І (така ситуація можлива при одній із форм спадкового порушення синтезу порфіринів). Наявність косинтази спрямовує конденсацію молекул порфобіліногену в бік утворення саме уропорфіриногенуІІІ.
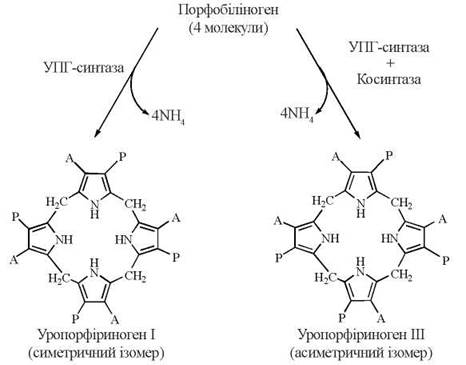
УПГ-синтаза — уропорфіриногенсинтаза; А — ацетат (-CH2COOH); Р — пропюнат (-CH2CH2COOH)
Уропорфіриноген ІІІ є також метаболічним попередником у синтезі вітаміну В12 бактеріями та хлорофілу рослинами і бактеріями.
4.2. Перетворення уропорфіриногену ІІІ на копропорфіриноген ІІІ. Механізм реакції полягає у декарбоксилюванні бічних ацетатних ланцюгів (фермент уропорфіриноген-декарбоксилаза).
4.3. Перетворення копропорфіриногену ІІІ на протопорфіриноген ІІІ та протопорфірин ІІІ (ІХ). Процес включає в себе окислювальне декарбоксилювання бічних ланцюгів та окислення метиленових мостиків і каталізується специфічними оксидазами мітохондрій — копропорфіриногеноксидазою та протопорфіриногеноксидазою, відповідно.
Включення в молекулу протопорфірину IX атома двовалентного заліза, що каталізується ферментом ферро-хелатазою (гем-синтазою) мітохондрій завершує синтез гему (протогему ІХ). Сполучення гему з білком глобіном призводить до утворення гемоглобіну.
Реакції перетворення уропорфіриногену ІІІ на молекулу протопорфірину ІХ та синтез гему:
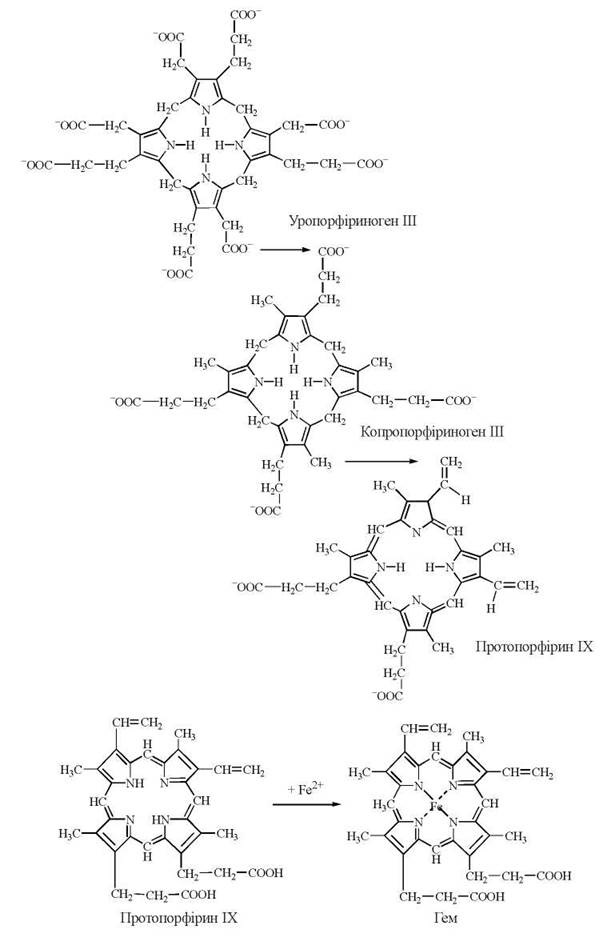
Джерелом іонів заліза гему є залізовмісний тканинний білок феритин, одна молекула якого може зв’язувати 4500 іонів Fe3+. В свою чергу, феритин отримує залізо від білка трансферту, що в плазмі крові транспортує залізо, яке надходить за рахунок розщеплення гемоглобіну еритроцитів та всмоктування іонів заліза в кишечнику.
Загальну послідовність реакцій синтезу гему з гліцину та сукциніл-КоА подано на схемі:
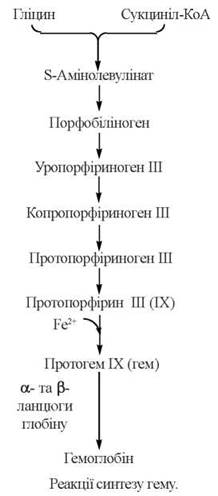
Регуляція синтезу порфіринів здійснюється на рівні ДАЛК-синтази за механізмом негативного зворотного зв’язку таким чином, що в умовах накопичення гему його власний біосинтез гальмується. Кінцевий продукт біосинтетичного шляху — гем — править за корепресор, що разом із білком апорепресором утворює негативний модулятор, який протидіє трансляції в рибосомах мРНК для ДАЛК-синтази, призводячи до блокування синтезу ферменту.
Спадкові порушення обміну порфіринів
Спадкові порушення біосинтезу порфіринів (порфіри) — дефекти метаболізму (ензимопати), за яких порфірини та їх попередники в надмірних кількостях накопичуються в тканинах людського організму, зокрема в шкірі і підшкірній клітковині, та екскретуються з сечею і фекаліями.
Існують декілька найбільш поширених клініко-біохімічних типів порфірій, що відрізняються типом дефектного гена та характером прояву ензимопатії. Описані порфірії, які розвиваються в результаті дефектів майже всіх ферментів синтезу гему. Успадковуються порфірії як автосомно-рецесивні або автосомно-домінантні хвороби.
Основними клінічними проявами порфірій є підвищення чутливості до світла та неврологічні порушення.
Світлочутливість
Аномальне відкладення порфіринів різної молекулярної структури в шкірі призводить до її фотосенсибілізації та розвитку фотодерматитів. Молекулярною основою таких патологічних проявів є утворення під дією сонячного світла з довжиною хвилі близько 400 нм активних форм кисню типу синглетного кисню 1О2та перекисних радикалів порфіринів типу RO2, які пошкоджують клітинні мембрани і призводять до загибелі клітин.
Неврологічні порушення
Неврологічні порушення при порфіріях проявляються патологічними симптомами з боку як периферичної (дизестезії, порушення моторики кишечника, нерво-м’язової провідності, параліч дихальних м’язів тощо), так і центральної нервової системи.
Залежно від основного місця прояву специфічного ферментного дефекту, розрізняють еритропоетичні та печінкові порфірії.
Еритропоетична порфірія (хвороба Гюнтера) — патологія, зумовлена порушенням синтезууропорфіриноген ІІІ-косинтази. В результаті цього біохімічного дефекту відбувається утворення нефізіологічного ізомера уропорфіриногену — уропорфіриногену І. Для захворювання характерна забарвленість в червоний колір сечі (в деяких випадках також кісток та зубів), зумовлена накопиченням нирками уропорфіриногену І, який у сечі перетворюється на уропорфірин І.
Печінкові порфірії. Розрізняють декілька типів печінкових порфірій, характерними проявами яких є неврологічні порушення, пов’язані з надмірним накопиченням в організмі серотоніну внаслідок зниження синтезу гемвмісного ферменту триптофанпіролази.
До найбільш поширених клінічних форм печінкових порфірій належать:
- гостра мінлива порфірія (піролопорфірія) — захворювання, зумовлене дефектом ферменту уропорфіриноген-синтази (порфобіліногендезамінази);
- спадкова копропорфірія — ензимопатія, спричинена дефектом ферменту копропорфіриногеноксидази.