Практическая химия белка - А. Дарбре 1989
Определение состава белковых олигомеров. Получение мономеров и полипептидных цепей
Идентификация мономеров
Методы выделения мономеров
Выделение и очистку мономеров с целью определения аминокислотного состава, получения пептидных карт или секвенирования осуществляют с помощью препаративных методов химии белка.
При работе с микроколичествами материала в белок вводят радиоактивную метку и пользуются аналитическими методами. Метка может быть внутренней (т. е. введенной in vivo) [14, 161] или присоединена химическими методами по функциональным группам аминокислот (как указано в разд. 1.4.5.1).
1.3.2.1. Разделение субъединиц по размерам. Если субъединицы заметно различаются по молекулярной массе, их выделяют с помощью гель-фильтрации в присутствии денатурирующих агентов. Основы и методика гель-фильтрации на аналитическом уровне рассмотрены в разд. 1.3.1.3; при фракционировании на препаративном уровне они существенно не меняются. Хроматографию, как правило, проводят на препаративных колонках диаметром 2,5:5 см и длиной 100 см, рассчитанных на нагрузку от 500 мг до нескольких граммов белка.
Для разделения используют легколетучие буферные системы, в частности разбавленные растворы аммиака, уксусную и муравьиную кислоты, что позволяет выделять белки из элюатов с помощью лиофильного высушивания. Препараты для аминокислотного анализа рекомендуется получать гель-фильтрацией в 50—75%-ной муравьиной кислоте. Муравьиная кислота, сильный денатурирующий и солюбилизирующий агент, вызывает разрушение носителей на основе полисахаридной матрицы; в частности, не рекомендуется оставлять сефадексы в контакте с 75%-ной муравьиной кислотой на срок более трех недель.
Колонки изготовляют из стекла, соединительные шланги — из тефлона (рис. 1.3). Белки выделяют из элюата с помощью лиофильного высушивания в вакуумной системе с ловушками, предварительно фракции разбавляют до концентрации муравьиной кислоты —10%. Принято считать, что муравьиная кислота не гидролизует пептидные связи, за исключением лабильной связи Asp-Pro.
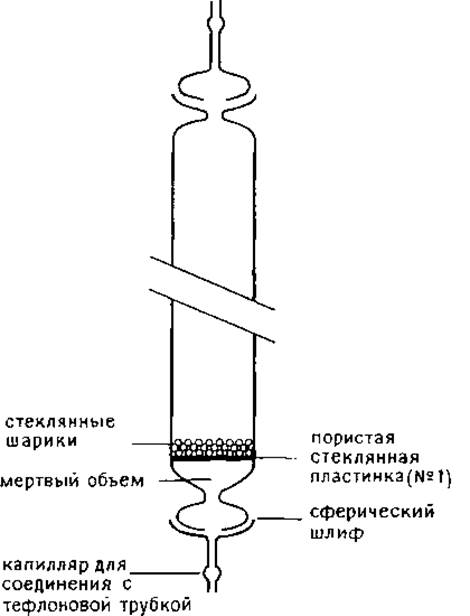
РИС. 1.3. Стеклянная хроматографическая колонка, предназначенная для проведения хроматографии с использованием в качестве элюента муравьиной кислоты.
При работе с количествами 2—3 мг и, возможно, до 100 мг определенным преимуществом перед традиционной гель-фильтрацией обладает ВЭЖХ. В этом случае укороченное время анализа вполне компенсирует небольшую емкость колонок. Несомненно, что благодаря технологическому прогрессу в недалеком будущем окажутся доступными колонки большой емкости, однако уже сегодня можно предполагать, что они будут иметь высокую стоимость. Мономеры можно разделять также с помощью препаративного электрофореза в ПААГ — ДСН. По производительности метод уступает гель-фильтрации: предельная нагрузка 50 мг белка, однако с помощью электрофореза можно разделять субъединицы с примерно равными молекулярными массами [168]. На практике в основном используются два различных варианта электрофореза. Первый вариант представляет собой модификацию аналитического метода. Можно, например, увеличить толщину пластины геля или, исключив гребенку, наносить образец в виде сплошной полосы. После проведения электрофореза для проявления белковых зон используют обработку по одной из следующих методик:
1. Гель выдерживают в красителе более короткое время или же используют более разбавленный раствор красителя (0,05%-ный раствор кумасси), чем это принято в типовой методике (разд. 1.2.1.1).
2. Окрашивают контрольные полоски геля (при сравнении с исходным гелем следует учитывать усадку пластины при обесцвечивании в смеси метанол — уксусная кислота).
3. Пропитывают гель 1 М КСl, после чего зоны белка обнаруживают по опалесценции за счет образования нерастворимого додецилсульфата калия [180].
4. Пропитывают гель 4 М ацетатом натрия, при этом зоны белка выявляются в виде прозрачных полосок (предел обнаружения 0,1 нг/мм3) [76].
Зоны, представляющие интерес, вырезают и белок выделяют экстракцией [49, 64] или элюированием в условиях электрофореза [2, 175]. При определении аминокислотного состава приготовленных таким способом образцов обязательно вводят необходимые поправки, учитывающие присутствие акриламидного геля [26]. Согласно другому варианту, белки разделяют электрофорезом в блоке акриламидного геля, при этом под действием электрического поля белковые фракции попадают в проточную камеру, откуда и вымываются раздельно потоком элюэнта. Ход элюирования регистрируют с помощью проточного денситометра, элюат собирают на фракционном коллекторе. Недавно разработана методика элюирования белков из системы ПААГ — ДСН в промежуточный гель с использованием восходящего электрофореза [121]. Выход достигается высокий, однако образец может содержать примеси компонентов акриламидного геля и буферной системы. Гели для препаративного электрофореза могут иметь форму столбиков (LKB) или пластин (блоков) (Biorad). Для разделений по описанной методике используют различную аппаратуру, выпускаемую фирмами.
1.3.2.2. Ионообменная хроматография. Для разделения белков на субъединицы по суммарному заряду используют методы ионообменной хроматографии, электрофореза (разд. 1.3.2.3), изоэлектрического фокусирования и хроматофокусирования (разд. 1.3.2.4).
При ионообменной хроматографии образец наносят на колонку с ионитом (сорбентом), несущим положительно (анионит) пли отрицательно (катионит) заряженные ионогенные группировки. На сорбенте, заряженном положительно, компоненты разделяемой смеси, также несущие положительный заряд (при условии, что pH буфера не превышает рK), проходят свободно с потоком элюента, а компоненты, заряженные отрицательно, удерживаются. Слабосорбированные вещества элюируют исходным буфером, сильносорбированные «снимают» с колонки, пропуская раствор с более высокой ионной силой или с другим pH. В результате компоненты смеси элюируются в порядке возрастания суммарного заряда.
Ионогенные группы фиксируют на различных инертных носителях; наиболее распространенные из них гранулированная целлюлоза (фирмы Whatman, Serva, Bio-Rad, Eastman) и сферическая целлюлоза (фирма Pharmacia). В качестве носителя используются также агароза и декстран (имеющие поперечную сшивку). Ионообменники на основе декстрана характеризуются высокой емкостью и с успехом применяются для разделения не очень крупных биомолекул. Главным недостатком ионитов этого типа является сильная зависимость объема гранул от ионной силы и pH буферного раствора. При разделении изоформ соевой липоксигеназы-1 методом ВЭЖХ с успехом использовался катионит TSK-Gel LS-212 [9]. Недавно фирма Pharmacia выпустила крупнопористый ионообменник для ВЭЖХ, позволяющий проводить разделение с высокой скоростью и хорошим разрешением (гл. 6).
Большинство анионитов в качестве ионогенных функциональных групп включают диэтиламиноэтильную (ДЭАЭ) или диэтил (2-гидроксипропил)аминоэтильную (ДГПАЭ) группы. В катионитах ионогенными группами являются карбоксиметильная (КМ), фосфатная (Ф) и сульфопропильная (СП) группы. На сильных ионообменниках (содержащие группы ДГПАЭ, Ф и СП) рекомендуется фракционировать вещества, проявляющие слабокислотные или слабоосновные свойства, или вести разделения в области экстремальных pH. Эффективность разделения белков па ионитах зависит не только от суммарного заряда. Так, белки с высоким содержанием ароматических аминокислот фракционировали на ДГПАЭ-целлюлозе при pH 0,5 по их гидрофобным свойствам [65].
Выбор ионообменника зависит от свойств исследуемых белков. Обычно хроматографию проводят в том буфере, при котором целевые компоненты смеси предельно различаются по величине суммарного заряда. При выборе рабочего диапазона pH можно руководствоваться результатами электрофореза или изоэлектрофокусирования (т. е. изоточками компонентов смеси) (разд. 1.3.2.4). При отрицательном суммарном заряде хроматографию проводят на анионите, при положительном суммарном заряде — на катионите. Выбору оптимальных условий ионообменного разделения должна предшествовать предварительная оценка свойств разделяемой смеси с помощью простых тестов.
Выбор ионообменника. Аликвотные части раствора белка смешивают с катионитом и анионитом, уравновешенными буфером с определенным pH. По оптической плотности жидкости над ионитом определяют степень связывания белка сорбентом и тем самым находят тип ионита и величину pH, при которых сорбция исследуемого белка достигает максимума. На практике в пробирки помещают по 0,05 г каждого ионита, например ДЭАЭ- и КМ-целлюлозу, и по 30 мл соответствующего буферного раствора (для анионита — 0,05 М трис-НСl, pH 8,5; для катионита — 0,05 М ацетат натрия, pH 5,0), содержащего 8 М мочевину (необходимую для диссоциации олигомера). Иониты приводят в равновесие с буферным раствором, заменяя супернатант свежим буфером дважды или более раз, большую часть жидкости над ионитом удаляют (так, чтобы общий объем ионита с буфером над ним составил 10 мл). В каждую пробирку прибавляют равное количество белка, перемешивают встряхиванием и после оседания ионита жидкость отделяют. Оптическую плотность супернатанта измеряют спектрофотометрически. Некоторые белки сорбируются как на катионите, так и на аниониге, в этом случае оба этих ионита пригодны в качестве хроматографического сорбента. В то же время, если белок не сорбируется па ДЭАЭ и КМ-целлюлозе, необходимо уменьшить концентрацию буфера или использовать более сильные иониты.
Методика проведения ионообменной хроматографии. Исчерпывающую информацию относительно свойств и физических характеристик различных ионитов, а также практические рекомендации по работе с ними можно найти в материалах, выпускаемых фирмами-изготовителями. Хороший обзор по целлюлозным ионообменникам опубликован Петерсоном [133].
Сухой ионит предварительно подвергают кислотной и щелочной обработке и лишь затем уравновешивают рабочим буфером. Аниониты, например ДЭАЭ-целлюлозу, суспендируют в 0,5 М НСl (15 мл/г) и перемешивают в течение 30 мин. Жидкость отделяют фильтрованием, ионит промывают водой и суспендируют при перемешивании в равном объеме 0,5 М NaOH в течение 30 мин. Жидкость вновь отделяют, ионит промывают водой. Затем ионит суспендируют в рабочем буфере (20 мл/г), перемешивают в течение 10 мин, отделяют путем декантации и фильтрованием. Операцию повторяют трижды до тех пор, пока pH и электропроводность жидкости над ионитом не сравняется с параметрами рабочего буфера. Катиониты обрабатывают аналогичным образом, однако в иной последовательности. Например, КМ-целлюлозу промывают вначале 0,5 М NaOH, а затем 0,5 М НСl. Иониты, поставляемые во влажном виде, не нуждаются в предварительной «тренировке», их уравновешивают рабочим буфером по обычной методике.
Далее удаляют мелкую фракцию ионита. Суспензию взмучивают в мерном цилиндре, оставляют в покое для отстаивания в течение 1 ч, жидкость отбрасывают.
Готовят суспензию, свободно пропускающую пузырьки воздуха, деаэрируют в вакууме водоструйного насоса и набивают колонку. Для препаративных целей используют колонки диаметром 1,5:2,5 см, длиной 50:100 см; перед набивкой колонку снабжают насадкой, позволяющей в один прием внести всю массу ионита. После набивки пропускают через слой ионита 1—2 объема рабочего буфера. Скорость промывания должна быть идентична предполагаемой скорости элюирования, а объем сорбента должен оставаться постоянным.
Образец растворяют в исходном буфере, в случае необходимости диализуют в течение 12 ч. Если целевые компоненты смеси сорбируются достаточно прочно, объем раствора образца значения не имеет. В случае слабой сорбции объем вводимого в колонку образца не должен превышать 1—2% ее объема. Нагрузка должна быть <10% емкости сорбента.
Ход элюирования регистрируют по изменению оптической плотности при 280 нм (или с помощью методов, описанных в разд. 1.2.2.1), элюат собирают по фракциям. Компоненты смеси, удерживаемые ионитом, элюируют, изменяя ступенчато или плавно ионную силу или pH рабочего буфера. Элюирование в ступенчатом градиенте приводит к появлению артефактов и вследствие этого применяется все реже.
Способы формирования градиентов. Градиент концентрации или градиент pH формируют с помощью двух сообщающихся сосудов или перистальтического насоса. Выпускаются также специальные формирователи градиента.
Система из двух сообщающихся сосудов с вертикальными стенками изображена па рис. 1.4, а. Обозначим площадь поперечного сечения смесителя 1 и резервуара 2 как А1 и A2, а концентрации исходного и конечного буфера С1 и С2. Если оба буферных раствора имеют одинаковую плотность, то изменение концентрации элюента можно описать следующим образом:
С = С2 — (С2 — С1) (1 — v/V)A2/A1
где V — объем элюэнта, поданного системой на колонку, V — суммарный объем смесителя и резервуара. При А1=А2 формируется линейный градиент, если А1<А2, градиент выпуклый, если А1>А2, градиент вогнутый [22].
Для построения градиентов можно использовать многоканальный перистальтический насос (рис. 1.4,6). Форма градиента определяется скоростью потока R1 (из сосуда 2 в сосуд 1) и R2 (из сосуда 1 в колонку), концентрациями C1 и С2 и объемом V1 сосуда 1. Через время t концентрацию буфера, поданного на колонку, можно вычислить по формуле [96]:
![]()
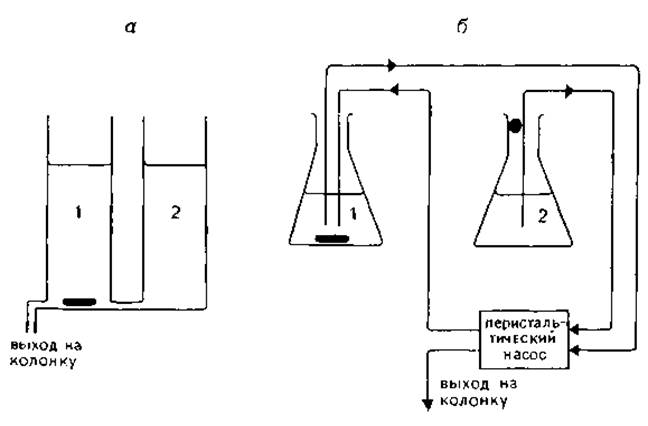
РИС. 1.4. Способы формирования градиента с помощью сообщающихся сосудов (а) и перистальтического насоса (б). 1 — смеситель; 2 — резервуар. Форма градиента определяется площадью поперечного сечения сосудов (а) или диаметром эластичных трубок перистальтического насоса (б).
Если R2 = 2R1, формируется линейный градиент, угол наклона которого определяется по формуле:
![]()
При R2>2R1 формируется выпуклый, а при R2<2R1 вогнутый градиенты. Если R1<R2, то соотношение объема смесителя V1 и суммарного объема элюента V должно удовлетворять следующему условию:
![]()
У большинства перистальтических насосов скорости потока R1 и R2 можно варьировать путем изменения диаметра трубок. Выпускаются насосы, позволяющие варьировать скорости потоков в широком диапазоне. Если диаметр трубок можно варьировать в ограниченных пределах, линейный градиент формируют с помощью трех каналов одинакового диаметра. По одному каналу подают буфер из сосуда 2 в сосуд а по двум остальным — буфер из сосуда 1 в колонку.
1.3.2.3. Электрофорез в полиакриламидном геле. С помощью аналитического электрофореза в присутствии 8 М мочевины идентифицируют мономеры, имеющие различные отношения отрицательного заряда к массе, а также делают заключение о типе попита и pH рабочего буфера для выделения мономеров методом ионообменной хроматографии. В препаративном варианте используются условия, повторяющие аналитическую систему, или электрофоретическое элюирование (разд. 1.3.2.1).
Выбору оптимальных условий электрофореза в ПААГ посвящена работа [33]. Имеется компьютерная распечатка 5000 буферных систем для электрофоретического разделения катионов и анионов в диапазоне pH 2,5—11 [35]. На практике используются лишь немногие из этих систем с наиболее типичными pH [119]. Далее описана система, предложенная Девисом [43], несколько модифицированная путем введения 8 М мочевины.
Часто возникает задача определения молекулярных масс белков, которые удалось электрофоретически разделить по величине заряда. В этом случае проводят электрофорез в трубке в присутствии 8 М мочевины, а затем в перпендикулярном направлении в блоке в присутствии ДСН (двумерный электрофорез в геле обсуждается в разд. 7.3.2).
Электрофорез при pH 8,9 в присутствии 8 М мочевины. Методика эксперимента, аппаратура, реагенты, красители, а также способы фоторегистрации описаны в разд. 1.2.1.1 и 1.3.1.1. Далее остановимся на модифицированной системе Девиса [43].
Буфер рабочего геля: З М трис, оттитрованный НСl до pH 8,9.
Буфер формирующего геля: 0,5 трис, оттитрованный фосфорной кислотой до pH 6,8.
Буфер для приготовления образцов; 2 мл буфера формирующего геля, 6 г мочевины, 5 мл воды, 5 мг бромофенолового синего.
Электродный буфер: 0,005 М трис + 0,04 М глицин.
Запасной раствор: 29,2% (масс./об.) акриламида, 0,8% (масс./об.) N,N'-метилепбисакриламида.
Соотношение компонентов для приготовления 10 мл рабочего акриламидного геля различной концентрации
|
7,5% |
10% |
12,5% |
|
|
30%-ный раствор акриламида |
2,50 мл |
3,33 мл |
4,17 мл |
|
Буфер для рабочего геля |
1,25 мл |
1,25 мл |
1,25 мл |
|
Мочевина |
4,8 г |
4,8 г |
4,8 г |
|
Вода |
2,65 мл |
1,82 мл |
0,98 мл |
|
ТЕМЕД |
5 мкл |
5 мкл |
5 мкл |
|
Персульфат аммония |
5 мг |
5 мг |
5 мг |
После прибавления к раствору персульфата аммония раствор деаэрируют под вакуумом водоструйного насоса и переносят в камеру для электрофореза (защищают от кислорода воздуха слоем воды или насыщенного водой изобутанола). По завершении полимеризации сливают защитный слой, поверхность промывают раствором формирующего геля и вносят предварительно деаэрированный раствор формирующего геля. Для приготовления 10 мл формирующего геля смешивают 1,3 мл запасного раствора, 1,25 мл формирующего геля, 4,8 г мочевины, 3,8 мл воды, 5 мкл ТЕМЕД, 5 мг персульфата аммония.
Образцы вносят с помощью микрошприца или микропипетки. Электрофорез проводят при 100 В до прохождения зоной красителя формирующего геля, а затем при 200 В до приближения зоны красителя к нижней границе геля.
Двумерный электрофорез. Проводят электрофорез в первом направлении при pH 8,9 (или ином pH) в присутствии 8 М мочевины, а затем во втором направлении в присутствии ДСН. С целью сужения зон па границе геля электрофорез во втором направлении проводят в ступенчатой буферной системе. Гель из трубки помещают па слон формирующего геля в торец пластины и фиксируют с помощью агарозы (концентрация ДСН в буфере для образца 1% (масс./об.), см. разд. 1.3.1) [115]. Электрофорез во втором направлении можно вести в непрерывной буферной системе па градиентном геле, тогда сужение зон будет идти вблизи стационарного положения. В этом случае гель из трубки (диаметр 3,5 мм) уравновешивают в течение 30 мин с 100 мл электродного буфера (0,025 М фосфат, pH 7 + 0,15% ДСН) и помещают на верхний торец готовой (фирменной) пластины градиентного геля (4—27%) (разд. 1.3.1.1), которую предварительно пропитывают ДСН с помощью электрофореза при 50 мА в течение 3 ч. Разделение проводят при 30 мА в течение 15 мин и 50 мА в течение 3,5 ч [114].
1.3.2.4. Изоэлектрофокусирование. При изоэлектрофокусировании раствор, содержащий смесь амфолитов-носителей (алифатических полиаминополикарбоновых кислот), подвергают воздействию электрического поля. При этом различные компоненты смеси мигрируют в ту область, где их суммарный заряд равен пулю, т. е. вещества с низкими изоэлектрическими точками двигаются в направлении анода. В результате между электродами формируется градиент pH. Амфолиты-носители выпускаются рядом фирм (LKB, Serva, Rio-Rad, Pharmacia, Pierce). Если в систему с градиентом pH, сформированным амфолитами, вносят белок, то образец мигрирует под воздействием поля в зону, соответствующую его изоэлектрической точке. Изофокусировапие позволяет наиболее точно оценить гетерогенность белков по заряду. Причину гетерогенности по заряду, о чем свидетельствует множество зон, выявить нелегко. Гетерогенность может быть обусловлена наличием в образце примесей других белков, химической модификацией белковых молекул вследствие дезамидирования, различной степенью фосфорилирования или взаимодействием цианата с N-концевой аминогруппой или ε-аминогруппой остатков лизина. Следовательно, предварительно необходимо охарактеризовать белок электрофоретически, а затем изучать с помощью изоэлектрофокусирования. Установлено, что из 500 исследованных белков 70% имеют изоэлектрические точки в кислой области, а 38% — в области pH 4,5—6,0 [63].
Аналитическое изоэлектрофокусирование в присутствии 8 М мочевины можно проводить в обычных гелях — полиакриламидном или сефадексе. Препаративное изоэлектрофокусирование проводят в колонках в градиенте плотности (сахарозы), однако в последние годы предпочтение отдают разделению в слое сефадекса или другого носителя (методики эксперимента описаны в буклетах фирм LKB, Pharmacia).
Вначале проводят изоэлектрофокусирование в широком диапазоне pH (например, 3:10), затем вторично — в более узком диапазоне (pH 5—7). Амфолиты-носители имеют высокую стоимость, поэтому в целях экономии разделение можно проводить вначале в трубках, а после тщательного подбора условий выполнять препаративное разделение.
Аналитическое изоэлектрофокусирование. Для приготовления 10 мл рабочего буфера смешивают 1,65 мл запасного раствора (30%-ного акриламида), 4,8 г мочевины, 3,75 мл воды, 1,0 мл раствора амфолитов, 5 мг персульфата аммония. Анодный буфер имеет состав: 0,03 М фосфорная кислота + 8 М мочевина; катодный буфер — 0,05 М NaOH + 8 М мочевина. Образец растворяют в буфере следующего состава: 1) 0,03 М фосфорная кислота + 8 М мочевина+ 20 %-ный глицерин (pH 4,0) или
2) 0,05 М трис + 8 М мочевина + 20%-ный глицерин (pH 8,0) (первый — в кислой и второй — в основной зоне геля). Проводят предварительное изоэлектрофокусирование при 400 В в течение 2 ч, наносят образцы, проводят разделение при 400 В в течение 17 ч. Гель промывают 10%-ной трихлороуксусной кислотой для удаления амфолитов, а затем проводят проявление (окрашивание). Проявление можно проводить прямым способом. Для этого гель пропитывают в течение 3 ч раствором следующего состава: 25%-ный этанол + 10%-ная уксусная кислота + 0,1 %-ный сульфат меди + 0,01 %-ный кумасси голубой R-250; раствор для обесцвечивания: 10%-ный этанол + 10%-ная уксусная кислота [144].
Градиент pH измеряют па неокрашенном геле с помощью контактного электрода (тип LoT 403-30, фирма Ingold). Отрезки проволоки вводят в гели с интервалом 1 см, измеряют pH, гель окрашивают и фотографируют [116]. Измерения проводят по крайней мере на трех гелях, из трех измерений находят среднее. Полученные значения изоэлектрических точек служат характеристикой белка в среде 8 М мочевины. Последнее замечание существенно, поскольку присутствие мочевины влияет на рК ионогенных групп [28, 176].
Молекулярные массы компонентов смеси можно определить с помощью двумерного электрофореза [128] по методике, описанной в разд. 1.3.2.3.
Препаративное изоэлектрофокусирование. Установка для электрофокусирования должна обязательно включать эффективно охлаждаемую горизонтальную пластину, электродные сосуды, источник питания.
15 г ультрадекса или сефадекса IEF (сефадекс G-75 после специальной обработки) выдерживают до полного набухания в 225 мл раствора 8 М мочевины, прибавляют 12 мл раствора амфолитов и тщательно перемешивают. Затем суспензию деаэрируют и помещают в специальную кювету (например, размером 200x200x5,0 мм) в охлаждаемом блоке, выравнивают суспензию стеклянной палочкой. Избыток влаги удаляют путем естественного испарения пли поглощают сухим сефадексом (открытую склянку с сефадексом накрывают марлей и фиксируют с помощью клейкой лепты па поверхности влажной суспензии). Подготовленный таким образом слой влажного геля при наклоне кюветы па 45 °С не должен смещаться.
Образец либо непосредственно наносят па поверхность геля (в виде полоски), либо после смешивания с сухим сефадексом (1 мл раствора образца в 8 М мочевине на 60 мг сефадекса) помещают в канавку, специально вырезанную в слое геля. Наносить образец можно в любой зоне рН-градиента, нагрузка составляет 1 мг белка на 1 мл суспензии геля.
Контакт слоя геля с электродным буфером осуществляется с помощью полосок бумаги ватман ЗММ, завернутых в целлофан или диализную пленку. В зависимости от расстояния между электродами и эффективности охлаждения электрофорез проводят при 300 В в течение 16 ч, а затем при 600 В в течение 4 ч. Белковые зоны обнаруживают методом отпечатков; для этой цели лист ватмана ЗММ помещают па слой геля и прикатывают роликом. Спустя две минуты лист бумаги снимают, высушивают, дважды промывают (10 мин) 10%-ной трихлоруксусной кислотой для удаления амфолитов, промывают (5 мин) раствором для обесцвечивания с целью удаления трихлоруксусной кислоты, окрашивают кумасси голубым G-250 (разд. 1.2.1.1) (10 мин) и обесцвечивают.
Зоны, содержащие исследуемые белки, извлекают, переносят в небольшую колонку (например, небольшой шприц с кусочком стекловаты), суспендируют в буфере. Амфолиты отделяют диализом или гель-фильтрацией на сефадексе G-10.
Иногда примеси амфолитов удаляются с трудом и мешают биологическому тестированию. В таких случаях следует испытать систему буффалит (Buffalyie), поставляемую фирмой Pierce [139]. Более детально методика работы обсуждается в ряде публикаций [140, 141] и в буклетах фирм-поставщиков.
1.3.2.5. Фракционное осаждение. Нейтральные соли оказывают специфическое и глубокое воздействие на конформационную стабильность белков [179], причем катионы и анионы, как правило, по-разному влияют на развертывание нативных структур. В то время как в присутствии небольших количеств солей растворимость белков возрастает (так называемый солевой эффект), при высоких концентрациях (обычно несколько молей) имеет место явление высаливания, которое и вызывает осаждение белков. Относительная эффективность нейтральных солей в реакции высаливания впервые была изучена в 1888 г. [79]. Предложены лиотропные ряды катионов и анионов:
![]()
Физико-химические основы явления высаливания белков в концентрированных растворах солеи достаточно сложны [163]. Одна из причин высаливания связывается со снижением активности ассоциированных с белком молекул воды, с их более слабым взаимодействием с полярными группами белка. К существенным факторам, определяющим растворимость белка, относят также поверхностное натяжение на границе между белковыми молекулами и на поверхности раздела белок — вода, причем лиотропный эффект объясняют влиянием посторонних ионов на величину поверхностного натяжения [27]. Растворимость 5 многих белков (при высокой солевой концентрации) уменьшается по мере увеличения концентрации соли по логарифмической зависимости:
lgS = ß — Ksw
где ß— растворимость белка в воде (находят обычно путем экстраполяции к w = 0), Ks— константа высаливания, w — ионная сила. Зависимости, приведенные на рис. 1.5, иллюстрируют изменение растворимости ряда белков в растворах сульфата аммония [36]. Как правило, наиболее полное осаждение белка наблюдается вблизи его изоэлектрической точки, однако это выполняется далеко не всегда.
Осаждение сульфатом аммония часто применяется в качестве предварительной стадии очистки белка, например с целью концентрирования или отделения от сопутствующих примесей. Иногда этот прием может оказаться полезным при разделении субъединиц, в частности если они имеют различную растворимость в сульфате аммония (или в растворах других солей).
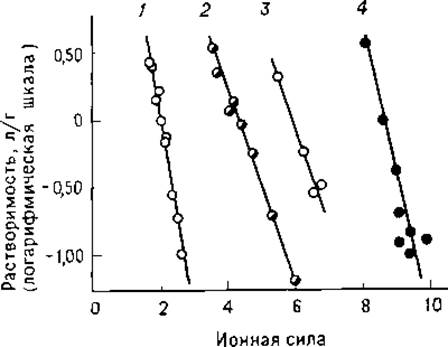
РИС. 1.5. Изменение растворимости белков в растворах сульфата аммония, 1 — фибриноген; 2 — гемоглобин; 3 — сывороточный альбумин; 4 — миоглобин [36].
Однако даже в благоприятных случаях осаждение приходится повторять, чтобы уменьшить возможное соосаждение других субъединиц. Сульфат аммония обладает несомненными преимуществами перед другими нейтральными солями: он имеет высокую высаливающую способность, не денатурирует белки, обладает низкой стоимостью и доступен в достаточно очищенной форме, хорошо растворим в воде, при растворении выделяется мало тепла. Кроме того, концентрированные растворы сульфата аммония имеют небольшую вязкость и плотность, что немаловажно, поскольку последующей стадией является ультрацентрифугирование.
На практике для достижения оптимальных результатов приходится действовать методом проб и ошибок. Вначале строят градуировочный график в определенной области концентраций сульфата аммония с учетом того, что сульфат аммония несколько снижает pH незабуференных растворов. Величина «процента насыщения» (100%-ное насыщение соответствует 4,05 М раствору при 20°С), характеризующая поведение конкретного белка, сильно варьирует в зависимости от свойств белка, его концентрации, pH (обычно pH 5,5—7,5) и температуры раствора (обычно 20°С). При приготовлении растворов сульфата аммония можно руководствоваться номограммой, приведенной в работе [48]. По номограмме легко определить количество соли, которое необходимо прибавить к определенному объему раствора для достижения нужной степени насыщения. В области рН<5 удается проводить осаждение белков при более низких концентрациях соли (такие растворы имеют более низкую плотность, что улучшает условия разделения фаз при центрифугировании).
Разделение с помощью органических растворителей. Белки можно фракционировать с помощью осаждения органическими растворителями, например этанолом, ацетоном. Белки, склонные к агрегации, осаждают сульфатом аммония в присутствии пропанола [51]. Органические растворители снижают растворимость белков благодаря изменению диэлектрической проницаемости среды. Поскольку причина осаждения состоит в агрегации молекул вследствие электростатического взаимодействия, что аналогично осаждению в изоэлектрической точке, pH в данном случае более важный фактор, чем при осаждении солями. Следовательно, с помощью органических растворителей можно разделить очень близкие в структурном отношении белки, имеющие разные изоэлектрические точки. Например, при pH 6,5 парвальбумины II, III и IV были разделены путем осаждения ацетоном [163]. Белок IV выпадал в осадок при концентрации ацетона 55—60%, белки III и IV — при концентрации 60—65%, белки III и II — при концентрации 65—70%, наиболее кислотные компоненты смеси с изоточками рК ∼ 4 выпадали в осадок при концентрации ацетона 70—80%.
Как правило, чем ниже молекулярная масса белка, тем выше концентрация органического растворителя, необходимая для его осаждения. Если концентрация растворителя слишком велика, осаждение может быть затруднено из-за белок-белкового взаимодействия. В этом случае в раствор добавляют биполярные вещества, например глицин, которые выполняют роль буфера и стабилизируют диэлектрическую проницаемость. Если белок денатурирует при комнатной температуре, осаждение проводят при пониженной температуре (при 0 °С). Прибавление к раствору белка органического растворителя вначале вызывает постепенное повышение температуры (до достижения 20%-ной (об./об.) концентрации), иногда уменьшение объема. Важно в процессе осаждения поддерживать постоянную температуру раствора, поскольку растворимость белков сильно снижается при охлаждении.
В этом отношении осаждение органическими растворителями отличается от фракционирования с помощью солеи, при котором зависимость растворимости от температуры не столь однозначна.
1.3.2.6. Прочие методы разделения. Здесь следует назвать прежде всего аффинную хроматографию, которая обсуждается в гл. 5.