Практическая химия белка - А. Дарбре 1989
Определение состава белковых олигомеров. Получение мономеров и полипептидных цепей
Идентификация мономеров
Методы определения молекулярной массы мономеров
Для определения молекулярной массы мономеров используют электрофорез в полиакриламидном геле (ПААГ) в присутствии ДСН и гель-фильтрацию в классическом и современном (ВЭЖХ) вариантах. Предпочитают использовать электрофорез в ПААГ или ВЭЖХ, однако в настоящее время ВЭЖХ уступает электрофорезу по качеству разрешения близких по молекулярной массе белков (гл. 6). Белки, содержащие более 10% углеводов, рекомендуется хроматографировать на агарозных гелях, так как электрофорез может не дать точных результатов из-за различия в связывании ДСН с полипептидной цепью и полисахаридными фрагментами. Следует также напомнить, что мономер может включать несколько полипептидных цепей, связанных дисульфидными связями, поэтому определение молекулярной массы следует проводить как до, так и после восстановления S—S-мостиков.
Молекулярную массу можно найти по данным количественного анализа N-концевых аминокислот (разд. 1.3.1.4). По расходу материала (количество образца) этот метод эквивалентен гель-фильтрации; при инструментальном анализе расход материала существенно меньше (гл. 12, 15, 17).
Статистическая обработка данных для 500 белков показала, что наиболее вероятная область молекулярных масс мономеров приходится на 10 000—60 000 [63]. Действительно, молекулярные массы субъединиц более чем 2/3 изученных к настоящему времени белков располагаются в указанном диапазоне. Примерно 50% этих белков составляют димеры, 30% — тетрамеры, 8% — гексамеры.
1.3.1.1. Электрофорез в ПААГ в присутствии ДСН, Большинство белков в присутствии ДСН диссоциируют на субъединицы. Субъединицы, состоящие из двух и более полипептидных цепей, связанных дисульфидными связями, могут подвергаться дальнейшей диссоциации в присутствии восстанавливающих агентов, например 2-меркаптоэтанола (разд. 1.5.1). Полипептидные цепи в общем связывают ДСН равномерно, примерно 1,4 г/г белка [143]; при этом комплекс приобретает сильный отрицательный заряд. Собственный суммарный заряд нативного белка маскируется, а отношение заряда к массе становится практически постоянным. Кроме того, ДСН вызывает конформационные изменения полипептидной цепи, хотя ее абсолютная конфигурация может быть и неизвестной (это может быть палочка, ожерелье, вытянутый эллипсоид, статистический клубок). Показано, что при электрофорезе в ПААГ комплексы мигрируют в соответствии с молекулярной массой [182]. Длину полипептидной цепи определяют путем сравнения электрофоретической подвижности комплекса с подвижностью белков-маркеров с известными молекулярными массами. На практике метод используется в трех вариантах:
1. Непрерывная буферная система (обычно фосфатный буфер [182]) в однородном геле.
2. Ступенчатая буферная система в однородном геле [95].
3. Непрерывная или ступенчатая буферная система в градиентном геле.
Каждому варианту свойственны собственные преимущества и недостатки. Общие принципы и методика эксперимента электрофореза в ПААГ подробно рассмотрены в ряде исчерпывающих публикаций [3, 35, 73, 183]. В ступенчатой системе образцы предварительно концентрируются в «формирующем» геле, после чего входят в рабочий гель в виде узких зон. В непрерывной системе предварительное концентрирование исключено и при нанесении образца в большом объеме по завершении электрофореза образуются широкие зоны. Если образец наносить в небольшом объеме, результаты в обеих системах должны быть идентичны. Наблюдающиеся иногда различия в результатах, возможно, обусловлены частичной диссоциацией комплекса при электрофорезе в ступенчатой системе.
По ряду причин многие белки связывают ДСН в аномально низком соотношении [164]. Причиной может быть необычный состав, присутствие углеводов; к этой группе относятся белки с высоким положительным или отрицательным зарядом. Электрофоретическая подвижность комплексов может меняться в зависимости от замен отдельных аминокислот, конформационных изменений в белках [40, 46, 126]. Подобное аномальное поведение можно обнаружить и свести к минимуму путем проведения электрофореза в гелях различной пористости и построения по результатам эксперимента графика Фергюсона (см. далее). Иными словами, для достоверной оценки кажущейся молекулярной массы в ступенчатой или непрерывной системе необходимо проводить измерение по крайней мере в четырех различных гелях.
При электрофорезе в градиентном геле полипептиды мигрируют до достижения зоны с определенной пористостью, препятствующей их дальнейшему продвижению. Если все комплексы полипептидов с ДСН имеют одинаковую форму, концентрацию ПААГ в этой зоне можно принять характеристической для данной молекулярной массы [97, 99, 101, 137]. Комплекс может частично разрушиться с потерей ДСН, в особенности в области, близкой к равновесному состоянию, однако это не влияет на конечный результат. Объем образца не влияет на ширину белковых зон, тем не менее наносить образец в большом объеме не рекомендуется. При электрофорезе в градиентном ПААГ формирование градиента с высокой воспроизводимостью может быть затруднительным. Однако для этих целей выпускаются преформированные гели высокого качества.
Аппаратура и реактивы. Полиакриламидные гели готовят в виде стержней в стеклянных трубках или в виде пластин; последний вариант более удобен при определении молекулярных масс. При электрофорезе в трубках различная электрофоретическая подвижность может быть обусловлена изменениями степени полимеризации геля, а также зависеть от длины слоя геля, условий проведения электрофореза. Техника извлечения геля из трубок разработана детально [35]; показано, в частности, что извлечение гелей с концентрацией мономеров более 10% бывает затруднительным. Рекомендации по использованию аппаратуры и меры безопасности при работе с реактивами обсуждаются в разд. 1.2.1.1.
30%-ный запасной раствор, содержащий 29,2% (масс./об.) акриламида и 0,8% (масс./об.) N,N'-метиленбисакриламида, фильтруют и хранят при 4°С (в этих условиях раствор можно хранить в течение нескольких месяцев). Перед употреблением раствор нагревают до 20°С и деаэрируют в вакууме водоструйного насоса.
N,N,N',N'-тетраметилендиамин (ТЕМЕД) перегоняют в вакууме и хранят в темном сосуде при 4 °С. Реактивы высокого качества поставляются фирмой Biorad и другими фирмами. Рекомендуется использовать ДСН («ос. ч.») фирмы BDH, поскольку на разрешение зон и общий конечный результат могут оказывать влияние примеси в ДСН, например натрийтетрадецилсульфат [21, 50, 118]. Остальные реактивы должны иметь квалификацию «ч. д. а.». Для дополнительной очистки ДСН можно перекристаллизовать из этанола (600 г растворяют в 3 л 80%-його этанола и обрабатывают активированным углем).
Полиакриламидные гели. Свойства геля зависят от общей концентрации мономеров (акриламид +N,N'-бисакриламид) и доли сшивающего агента (N,N'-метиленбисакрил амида) [34]. Гели готовят путем радикальной полимеризации мономеров, в водном растворе в присутствии катализатора — персульфат аммония + ТЕМЕД.
После внесения персульфата раствор перемешивают до растворения кристаллов и быстро переносят в систему для проведения электрофореза. Следует принять меры против образования в геле пызырьков воздуха. Для этого на иглу шприца надевают отрезок тонкого шланга (силиконового, полиэтиленового), по которому осторожно по стенке наслаивают рабочий раствор мономеров. Гребенку для формирования лунок осторожно погружают в раствор, удаляют с зубцов пузырьки воздуха и выдерживают раствор до завершения полимеризации. Скорость полимеризации может варьировать в зависимости от концентрации персульфата аммония и ТЕМЕД. В среднем полимеризация должна завершиться спустя 10—25 мин после добавления персульфата. Ускоренная или замедленная полимеризация плохо воспроизводится и может привести к образованию геля с неравномерными порами.
Соотношение компонентов для приготовления 10 мл акриламидного геля различной концентрации
|
5% |
7,5% |
10% |
12,5% |
|
|
30%-ный раствор акриламида |
1,67 мл |
2,50 мл |
3,33 мл |
4,17 мл |
|
Запасной буфер (фосфат-ДСН) |
2,0 мл |
2,0 мл |
2,0 мл |
2,0 мл |
|
Вода |
6,33 мл |
5,50 мл |
4,67 мл |
3,83 мл |
|
ТЕМЕД |
5 мкл |
5 мкл |
5 мкл |
5 мкл |
|
Персульфат аммония |
10 |
10 |
10 |
10 |
Приготовление образца. Лиофильно высушенный образец белка растворяют в стартовом буфере (см. далее), образец в растворе диализуют против стартового буфера в течение 3—12 ч. Равновесная концентрация ДСН в образце достигается во времени, поэтому перед диализом к раствору белка добавляют сухой ДСН [до концентрации 1% (масс./об.)]. С целью полноты денатурации и связывания с ДСН образцы инкубируют при 95—100°С в течение 2 мин. Содержание белка в образце должно быть 0,5—1 мкг/мкл; в зависимости от толщины геля в лупки вносят 2—10 мкл такого раствора. Нагрузка существенно меньше, если обнаружение предполагают вести с помощью солей серебра. При проведении электрофореза в ступенчатой системе можно в случае необходимости вносить и большие объемы, при работе в непрерывной системе разбавленный образец концентрируют. С этой целью белок осаждают сульфатом аммония или трихлороуксусной кислотой, осадок вновь растворяют в стартовом буфере до достижения оптимальной концентрации (0,5—1 мкг/мкл).
Если белок плохо растворим (имеются в виду липопротеины и мембранные белки), в образец добавляют 8 М мочевину или неионный детергент, например 1%-ный тритон X-100, или увеличивают концентрацию ДСН до 10%. В последнем случае образец и буферные растворы не должны содержать ионов калия или солей гуанидина, так как эти анионы образуют с ДСН плохо растворимые соли. Полиакриламидные гели градуируют с помощью белков-маркеров, наборы которых выпускаются рядом фирм (Pharmacia, Biorad, BDH, Sigma, Calbiochem).
Непрерывная буферная система с постоянной концентрацией полиакриламида [182]. Основные компоненты этой системы: 0,05 М фосфатный буфер и 0,1%-ный ДСН.
Запасной буфер имеет следующий состав: 0,25 М фосфат (pH 7)+0,5% ДСН + 0,02% азид натрия (13 г NaH2PО4∙2H2О + 26 г Na2HPO4 + 0,2 г NaN3 + 5,0 г ДСН). Для растворения фосфатов необходимо слабое нагревание; хранят раствор при температуре выше комнатной.
Электродный буфер готовят из запасного раствора путем разбавления дистиллированной водой в соотношении 1 :4.
Образцы растворяют в буфере следующего состава: 20 мл запасного буфера + 1 г ДСН + 10 г глицерина + 0,05 г бромофенолового синего + дистиллированная вода (до объема 100 мл). При электрофорезе с целью определения молекулярной массы полипептидных цепей с восстановленными дисульфидными связями буфер должен содержать 1% (об./об.) 2-меркаптоэтапола.
После кратковременного нагревания до кипения образцы вносят в лунки геля с помощью микрошприца или микропипетки. Электрофорез ведут при 200 В до подхода зоны красителя к нижнему краю геля. Продолжительность эксперимента зависит от длины слоя геля и геометрии пластины. По завершении электрофореза проводят операции окрашивания, обесцвечивания и фотографирования (разд. 1.2.1.1).
Ступенчатая система с постоянной концентрацией полиакриламида [95]. Основные компоненты этой системы 0,375 М трис и 0,1 %-ный ДСН.
Концентрация акриламида в рабочем геле 5%—25%, в формирующем геле (длиной 20 мм) 4,5%.
Рабочий буфер имеет следующий состав: 1,5 М трис + 0,4% (масс./об.) ДСН, pH 8,8 (подтитрован НСl до указанного pH).
Буфер формирующего геля имеет следующий состав: 0,5 М трис + 0,4% (масс./об.) ДСН, pH 6,8 (подтитрован с помощью НСl).
Образец растворяют в буфере следующего состава: 0,0625 М трис + 2,3% (масс./об.) ДСН + 10% (масс./об.) глицерина + 0,05% (масс./об.) бромофенолового синего.
При определении молекулярной массы полипептидных цепей с восстановленными дисульфидными связями буфер содержит 1% (об./об.) 2-меркаптоэтаиола или 100 ммоль/л дитиотреита.
Электродный буфер имеет следующий состав: 0,025 М трис + 0,192 М глицин + 0,1 % (масс./об.) ДСН.
Соотношение компонентов для приготовления 10 мл акриламидного рабочего геля различной концентрации
|
5% |
7.5% |
ю% |
12,5% |
|
|
30%-ныи раствор акриламида |
1,67 мл |
2,50 мл |
3,33 мл |
4,17 мл |
|
Буфер для рабочего геля |
2,5 мл |
2,5 мл |
2,5 мл |
2.5 мл |
|
Бода |
5,83 мл |
5,00 мл |
4,17 мл |
3,33 мл |
|
ТЕМЕД |
5 мкл |
5 мкл |
5 мкл |
5 мкл |
|
Персульфат аммония |
10 мг |
10 мг |
10 мг |
10 мг |
Раствор деаэрируют дважды — перед добавлением ДСН и (вторично) перед прибавлением персульфата и ТЕМЕД. После внесения в систему па поверхность раствора осторожно наслаивают воду или насыщенный водой изобутанол. По завершении полимеризации верхний слой отсасывают, промывают поверхность геля буфером формирующего геля и наслаивают раствор для приготовления формирующего геля. Для приготовления 10 мл этого раствора (концентрация 4,5%) смешивают 1,5 мл запасного буфера, 2,5 мл буфера формирующего геля, 6,0 мл воды, 10 мг персульфата аммония, 10 мкл ТЕМЕД.
После кратковременного нагревания до кипения образцы вносят в лунки с помощью микрошприца или микропипетки. Электрофорез проводят при 100 В до прохождения красителем формирующего геля, затем при 200 В до подхода зоны красителя к нижнему краю геля. Продолжительность электрофореза зависит от длины геля и геометрии камеры, обычно электрофорез длится 3 ч.
Непрерывная или ступенчатая система в градиентном полиакриламиде. В этой методике используются рассмотренные ранее основные буферные системы. Единственное отличие от предшествующих систем состоит в том, что в блоке формируется градиент концентрации акриламида. Градиент образуют с помощью смесителя из двух сообщающихся сосудов и перистальтического насоса [113] или смесителя иной конструкции.
Фирмы Gradipore и Pharmacia производят преформированные пластины высокого качества (разд. 1.2.1.1). Преформированные гели не содержат ДСН, детергент вводят в гель на стадии предварительного электрофореза при 50 мА в течение 3 ч. Для приготовления электродного буфера запасной раствор (содержащий фосфат и ДСН) разбавляют дистиллированной водой в соотношении 1:9. После внесения образцов проводят электрофорез при 50 мА в течение 3,5 ч. За это время белковые зоны достигают равновесного положения, ограниченного размерами пор.
1.3.1.2. Анализ полученных результатов. Как отмечалось в разд. 1.2.1, молекулярные массы полипептидных цепей определяют с помощью градуировочного графика. Однако при электрофорезе в геле с постоянной концентрацией (пористостью) не удается исключить ошибки, обусловленные различием свободной подвижности Y0 исследуемого белка и белков-маркеров [148, 149]. Поэтому рекомендуется проводить электрофорез в нескольких гелях различной концентрации. При этом результаты определения молекулярной массы будут тем точнее, чем меньше систематическое отклонение при изменении общей концентрации акриламида. Полученные результаты можно проанализировать с помощью эмпирического уравнения Фергюсона [57]
lgRt = -KRT + lgY0
Коэффициент удерживания Kr для исследуемого белка и белков-маркеров определяется по наклону графика в координатах lgRf — общая концентрация акриламида Т, а молекулярную массу исследуемого белка находят по графику KR — молекулярная масса белков-маркеров. Для надежного определения KR электрофорез следует проводить по крайней мере в четырех гелях различной концентрации (7, 8; 9 и 10%), но предпочтительнее в семи гелях. Результаты экспериментов обрабатывают методами статистического анализа [148].
1.3.1.3. Гель-фильтрация. Общие принципы гель-фильтрации нативных белков рассмотрены в разд. 1.2.2; хроматография в денатурирующих условиях не имеет существенных особенностей. В присутствии денатурирующих агентов (гуанидингидрохлорид, мочевина, ДСН) меняется конформация белковых молекул, что часто приводит к увеличению гидродинамического объема. Поскольку разделение идет по размерам молекул, меняются диапазон молекулярных масс, а также градуировочный график для конкретного геля. Например, в неденатурирующих условиях на сефарозе CL-6B можно разделить белки с М 104—4∙106; в денатурирующих условиях в присутствии гуанидингидрохлорида диапазон молекулярных масс составляет 3000—80 000 [8].
Показано, что при высокой концентрации гуанидин-HCl восстановленные белки приобретают конформацию статистического клубка. В качестве эмпирического метода определения молекулярных масс денатурированных белков рекомендуется использовать хроматографию в 6 М гуанидин-HCl на гелях агарозы,
не сшитых поперечными связями (сефарозе, ультрогеле, биогеле А) [60, 111]. Главными недостатками этого метода являются продолжительность процесса (3—5 дней) и плохая устойчивость несшитой агарозы в присутствии 6 М гуанидин-НСl [110]. Сшитые агарозные гели (например, сефароза CL-6B) более устойчивы; в присутствии гуанидин-HCl свойства таких гелей остаются неизменными по крайней мере в течение года, а скорость элюирования может быть увеличена в 3—4 раза [8, 25, ПО]. Гель-фильтрация в присутствии гуанидин-HCl особенно важна при анализе гликопротеинов, поскольку в случае высокого содержания в белке углеводов электрофорез в ПААГ- ДСН дает ненадежные результаты. Хроматография на сефакриле S-200 — эффективный метод определения молекулярных масс небольших полипептидов с M > 30 000 [18]. К качеству гуанидин-НСl предъявляются высокие требования; рекомендуется использовать препарат квалификации «ос. ч.» фирмы Schwarz-Mann. В случае необходимости гуанидин-НСl очищают перекристаллизацией или готовят из гуанидинкарбоната [127].
При работе па большинстве гелей в качестве компонентов элюирующего буфера используется также 8 М мочевина или 0,1% ДСН. Показано, однако, что несшитые агарозные гели медленно разрушаются при высоких концентрациях мочевины. Элюирующий буфер (0,05 М фосфатный или 0,1 М аммонийбикарбонатный) обычно имеет pH 7—8. При использовании мочевины в буфер добавляют 0,1 М хлорида натрия с целью уменьшить электростатическое взаимодействие между матрицей геля и заряженными группами полипептидной цепи. Обнаружено [47], что в водных растворах мочевины самопроизвольно образуется цианат:
![]()
Накопление цианата идет с максимальной скоростью при pH >6, однако заметные количества цианата образуются и в кислой среде, даже при 0°С [71]. Кинетические исследования реакции цианата с аминогруппой белка показывают, что цианат и аминогруппа вступают в реакцию в непротонированной форме согласно уравнению
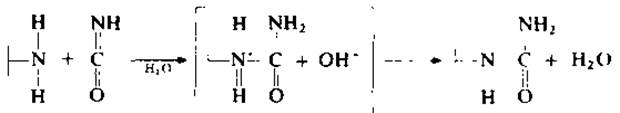
Это согласуется с различиями в скорости реакции а-аминогруппы (рКА≈8) и ε-аминогруппы (рКА ≈ 10,7): при pH 7 скорость реакции а-аминогруппы в 100 раз выше [166]. В области рН<4 и рН>10 карбамилирование аминогрупп не идет [174]. Помимо аминогрупп циана г вступает в реакцию с тиольными, карбонильными, имидазольными, фосфатными и реакционноспособными гидроксильными группами белков [166].
Здесь уместно напомнить, что интактные дисульфидные связи могут продолжать удерживать полипептидную цепь в свернутом состоянии даже при денатурации в 6 М гуанидин-HCl. В результате гидродинамический объем полипептидной цепи оказывается уменьшенным по сравнению с состоянием полной денатурации (с разрывом S—S-связей). Этот эффект становится более заметным по мере удлинения полипептидной цепи, увеличения числа дисульфидных групп и размеров петель. В таких случаях следует проявлять известную осторожность в оценке результатов определения молекулярных масс. С целью контроля проводят хроматографический анализ после восстановления и алкилирования дисульфидных мостиков. Сумма молекулярных масс отдельных полипептидных цепей должна соответствовать общей молекулярной массе исследуемого белка. Если после восстановления S—S-связей стадию алкилирования опускают, хроматография должна проводиться при pH 3 (в условиях, когда SН-содержащие цени достаточно устойчивы при комнатной температуре) пли в присутствии восстановителя, например
2-меркаптоэтапола (0,1 моль/л). Присутствие восстанавливающего агента часто мешает определению белков в элюате при 280 нм, поскольку в этой области поглощает окисленная форма 2-меркаптоэтанола, В таком случае применяют другие методы анализа, например перед хроматографией в белок вводят радиоактивную метку (разд. 1.4.5.1).
Получение гуанидингидрохлорид а [127], 1 кг гуанидинкарбоната растворяют при 40 °С в 2 л воды, прибавляют 3 л этанола и выдерживают на холоду в течение нескольких часов. Кристаллическую массу отделяют фильтрованием, дважды промывают водным этанолом (этанол : вода = 2 : 1), затем этанолом. Перекристаллизованный гуанидинкарбонат (выход 880 г) растворяют в 500 мл воды, раствор охлаждают на ледяной бане и при перемешивании медленно прибавляют к суспензии 6 М НСl до достижения в растворе pH 4 (эта величина pH должна сохраняться в течение 12 ч). Полученный гуанидингидрохлорид очищают путем дробной кристаллизации. Часть раствора упаривают при 40 °С до начала кристаллизации, выдерживают на холоду, кристаллы отделяют фильтрованием, маточный раствор объединяют с очередной порцией основного раствора п повторяют указанную последовательность операций. Конечный продукт — 780 г гуанидингидрохлорида — может быть вторично перекристаллизован из воды по описанной методике или из метанола. В случае необходимости для удаления окрашенных примесей используют активированный уголь. Оптическая плотность 6 М раствора гуанидингидрохлорида при 225 нм должна составлять 0,1.
Приготовление растворов мочевины. Как уже отмечалось, при хранении растворов мочевины образуется цианат, причем скорость образования цианата зависит от pH раствора [71]. Поэтому непосредственно перед использованием растворы мочевины обязательно надо пропускать через колонку со смешанным ионообменником (например, через колонку с элгалитом). Контроль раствора мочевины осуществляют путем измерений электропроводности, которая должна иметь величину ≤2 мкСм. Для связывания цианата в растворы мочевины иногда добавляют амины, например трис [75] пли этанол- амин [41].
1.3.1.4. Количественный анализ N-концевых аминокислот. Определение N-концевых аминокислот используется для доказательства высокой степени очистки олигомера (мономера) или отдельных полипентидных цепей. По данным количественного анализа N-концевых аминокислот можно получить представление о молекулярной массе исходного белка или рассчитать число полипептидных цепей. Например, если содержание N-концевой аминокислоты окажется в 2 раза выше теоретического или будут обнаружены две различные аминокислоты в эквимолярном соотношении, можно сделать вывод, что олигомер состоит из двух различных пептидов. При этом предполагают, что N-концевые аминокислоты не заблокированы, например из-за карбамилирования в растворах мочевины. Некоторые аминокислоты, например аспарагин, глутамин, серии, неустойчивы в условиях деградации по Эдману; в этом случае при интерпретации результатов вводят поправки.
Обычно используют прямой вариант метода Эдмана, ФТГ- аминокислоты определяют количественно по поглощению при 269 нм в этаноле [55]. При количественном определении ФТГ-аминокислот рекомендуется провести предварительную очистку модифицированного белка, а затем гидролиз и экстракцию.
Разработана методика, где метод изотопного разбавления используется в сочетании с деградацией по Эдману [15]. Согласно одному из вариантов этого метода, проводят одну стадию деградации по Эдману, в ходе которой блокируются ε-аминогруппы остатков лизина, а затем конденсируют второй N-концевой остаток с реагентом, содержащим радиоактивную метку [94]. В этом случае пет необходимости экстрагировать производное N-копцевой аминокислоты. Для количественного определения белка в образце используют аминокислотный анализ, а в аликвотных пробах гидролизата определяют радиоактивность. Для определения удельной радиоактивности [14С]ФИТЦ образец очищенного пептида модифицируют аналогичным образом.
С целью повышения растворимости белка па первой стадии можно использовать сульфофенилизотиоциаиат [23] или на второй стадии ввести в буфер ДСН (1%). В случае пенообразования из-за присутствия ДСН на стадии экстракции бензолом добавляют каплю н-октанола.
Количественное определение N-концевых аминокислот [15]. Количество белка в образце определяют с помощью аминокислотного анализа. Точную навеску (W в миллиграммах) лиофильно высушенного обессоленного белка конденсируют с ФИТЦ с известной удельной радиоактивностью, удаляют избыток реагента, проводят стадии отщепления и циклизации. Полученную таким путем меченую ФТГ-аминокислоту смешивают с известным количеством (N, в молях) немеченой ФТГ-аминокислотой и определяют удельную радиоактивность. Молекулярную массу Мr субъединицы рассчитывают по формуле
![]()
S1 и S2 (Ки∙моль-1)—удельная радиоактивность ФИТЦ и ФТГ-аминокислоты соответственно.
Деградация по Эдману. К аликвотной части (400 мкл) раствора белка (~10 мг/мл) известной концентрации прибавляют 600 мкл пиридина, 47 мкл N-диметилаллиламина и ~3 мкл безводной трифтороуксусной кислоты, pH реакционной смеси 9,5. Затем добавляют 10 мкл фенил-15С-изотиоцианата известной удельной радиоактивности и смесь инкубируют в атмосфере азота при 40 X в течение 1,5 ч. Избыток реагента экстрагируют бензолом (2 мл х З), водную фазу высушивают лнофильно. К сухому остатку прибавляют 1 мл воды и ледяную уксусную кислоту, насыщенную сухим HCl, инкубируют при 40 °С в течение 2 ч. (Методику проведения деградации по Эдману см. также в гл. 12.)
Выделение и идентификация ФТГ-аминокислоты. К полученной реакционной смеси прибавляют известное количество немеченого производного N-концевой аминокислоты, экстрагируют этилацетатом (2 мл х 2), экстракты объединяют и высушивают досуха. ФТГ-аминокислоту выделяют с помощью хроматографии на бумаге ватман SG81 в двух системах растворителей. Вначале используют систему хлороформ — метанол (9:1); зону, соответствующую положению ФТГ-аминокислоты, элюируют этилацетатом. Вторично хроматографируют в хлороформе, зону экстрагируют этанолом. Выделять ФТГ-аминокислоты можно также с помощью ВЭЖХ (гл. 13). Концентрацию ФТГ-аминокислоты в этаноле определяют, измеряя оптическую плотность 1 при 269 нм (ε = 16 000 М-1∙см-1) против этанола. Степень чистоты определяют по отношению А245/А269 (для эталона это отношение равно 0,39). Для определения радиоактивности отбирают аликвотные части (~3 мкл).
Определение удельной радиоактивности фенилизотиоцианата. Меченый ФИТЦ (0,5 мКи) разбавляют немеченым реагентом до удельной радиоактивности 0,4 Ки/моль. Аликвотную часть (2 мкл) реагента прибавляют к образцу N-концевой аминокислоты (2 мг) в 1 мл водного пиридина (1:1) и инкубируют в атмосфере азота при 50 °С в течение 35 мин. Избыток ФИТІI, экстрагируют бензолом (2 млХ4), водную фазу высушивают лиофильно. Циклизацию и перегруппировку в фенилтиогидантоин проводят путем инкубации в 1 М НСl (500 мкл) при 80 °С в течение 10 мин. ФТГ-аминокислоту экстрагируют этилацетатом (2 млХ2), а затем очищают хроматографией на бумаге ватман SG81 или с помощью ВЭЖХ. Содержание ФТГ-аминокислоты определяют по оптической плотности при 269 нм, затем измеряют число импульсов в аликвотной части и рассчитывают удельную радиоактивность. Среднюю удельную радиоактивность рассчитывают на основании анализа четырех образцов ФТГ-аминокислоты.
Сцинтилляционные жидкости. Для приготовления жидкого сцинтиллятора 5 г 2-(4-трет-бутилфенил)-5-(4-бифенил)окса-3,4-диазола (бутил-ФБД) растворяют в 5 л толуола (высушенного над натриевой проволокой). Образцы ФТГ-аминокислоты в 3 мл этанола смешивают с 15 мл жидкого сцинтиллятора н регистрируют на счетчике радиоактивности до накопления 40 000 имп. Радиоактивность образца рассчитывают по формуле Dn = Nн/E, где Nн — наблюдаемое число импульсов, а Е — эффективность счета (для данного сцинтиллятора и счетчика радиоактивности). Е находят по градуировочному графику, E = Nc/Dс, где Nc имп./мин (регистрируется прибором), a Dc — расп./мин эталона. Эффективность счета ß-излучения радиоактивного атома углерода составляет 92—95%.