БИОХИМИЯ И МОЛЕКУЛЯРНАЯ БИОЛОГИЯ - В. ЭЛЛИОТ - 2002
ГЛАВА 24. КЛОНИРОВАНИЕ ГЕНА, МЕТОД РЕКОМБИНАНТНОЙ ДНК, ГЕННАЯ ИНЖЕНЕРИЯ
Практическое использование достижений в области биохимии в данной книге специально не рассматривается, однако методы, в основе которых лежат манипуляции с ДНК, произвели поистине революционные изменения в биологии и особенно в биохимии. Поэтому мы посчитали целесообразным посвятить современным методам молекулярной биологии отдельную главу.
Благодаря им стало возможным выделение отдельных генов, определение в них последовательности оснований, а также перенос генов от одного вида организмов к другому и т. д. Зная последовательность оснований кодирующей области, можно определить аминокислотную последовательность белка, соответствующего данному гену.
Выяснение принципов функционирования и механизмов регуляции гена невозможно без данных о его структуре. Большая часть информации о регуляции активности гена, приведенной в главе 21, была получена в экспериментах с использованием рекомбинантной ДНК.
Изолированный ген может быть включен во множество различных типов клеток для продуцирования любых количеств кодируемого этим геном белка. Рассматриваемые в данной главе методы находят свое применение и в медицинской диагностике генетических заболеваний.
В чем заключались проблемы выделения генов?
Прежде всего - в гигантском размере молекул ДНК хромосом. В диплоидной эукариотической клетке каждый гомозиготный ген представлен двумя копиями - по одной на каждую из пары гомологичных хромосом. Среди тысяч других генов они образуют фракцию общей ДНК клетки.
Единственное, что отличает каждый конкретный ген от остальной ДНК, это закодированная в последовательности его оснований информация. Задача выделения гена сопоставима даже не с поиском иголки в стоге сена, а с поиском определенного пучка сена в этом стоге! Не нужно быть слишком большим пессимистом, чтобы оценить «тупиковость» ситуации. Все изменила технология рекомбинантной ДНК.
Первый шаг: разрезание ДНК эндонуклеазами рестрикции
Первый шаг при выделении гена заключается в разрезании клеточной ДНК на определенные, достаточно малые фрагменты, с которыми можно дальше работать. Эта цель сама по себе казалась недостижимой. Единственным ферментом, гидролизующим фосфодиэфирные связи в ДНК (см. с. 231), была ДНКаза, обнаруженная среди пищеварительных ферментов панкреатического сока. Если ДНК инкубировать с таким ферментом, произойдет фрагментация молекулы, однако разрыв связей происходит беспорядочно.
Это объясняет, почему открытие другого класса эндонуклеаз, или ДНКаз, у бактерий имело революционное значение: эндонуклеаза атакует внутренние связи в молекуле, а не концевые, как экзонуклеаза. Новый класс ферментов называют ферментами рестрикции, а вызываемые ими разрывы - рестрикцией. О молекуле ДНК, обработанной такими ферментами, говорят, что она была подвергнута рестрикции. Эндонуклеазы рестрикции гидролизуют ДНК не беспорядочно: каждый узнает свою короткую специфическую последовательность оснований и производит разрез в точно установленном месте. У различных бактерий имеются ферменты рестрикции, узнающие разные последовательности в ДНК и, следовательно, осуществляющие разрезы в разных участках (сайтах рестрикции). Например, фермент из Е. coli разрезает двуцепочечную ДНК у последовательности:
![]()
А фермент из Bacillus amyloliquefaciens специфичен в отношении другого сайта рестрикции:
![]()
Запоминать эти последовательности не стоит, но обратите внимание, что они имеют двойную симметрию, т. е. при чтении обеих цепей в направлении 5' —> 3' последовательности оснований идентичны. Ферменты получают свое название по имени бактерий (или бактериальных штаммов), из которых они выделены, а римская нумерация используется в тех случаях, когда штаммы бактерий имеют несколько таких ферментов. Вышеупомянутые ферменты названы соответственно ЕсоRI и ВаmНI. ЕсоRIбыл первым ферментом, выделенным из R-штамма Е. coli. Другие ферменты рестрикции узнают последовательности из 4, 5 и 8 оснований. Ферменты рестрикции делают возможным разрезание ДНК в строго установленных местах, определяемых последовательностями оснований, с хирургической точностью, образуя заданные фрагменты.
Какова биологическая функция ферментов рестрикции?
Ферменты рестрикции в бактериальных клетках предназначены для разрушения чужеродных ДНК. Например, бактериофаг, или фаг лямбда (λ), инъецирует свою ДНК в клетку Е. coli (см. с. 314). Фермент рестрикции разрезает эту ДНК в сайтах рестрикции, чем предупреждает успешное размножение фага и не дает возможности чужеродной ДНК инфицировать клетку. Почему фермент не разрушает собственную ДНК клетки? Гексамерная последовательность оснований, подобная распознаваемой EcoRI, должна встречаться в хромосоме Е. coli много раз; статистически - каждые 46 (4096) пар оснований. Клетка защищает от разрезания собственную ДНК, добавляя сразу же после ее синтеза метильную группу во все последовательности обеих цепей, узнаваемые ферментом рестрикции. Это не мешает спариванию оснований и экспрессии гена, но фермент рестрикции не узнает метилированную последовательность, а, следовательно, собственная ДНК клетки нечувствительна к атаке этого фермента.
Биологическая роль ферментов рестрикции может быть проиллюстрирована на примере фага А, инфицирующего клетки Е. coli. Разные штаммы Е. coli различаются по ферментам рестрикции, поэтому фаг А, который уже реплицировался в одном штамме Е. coli, будет инфицировать его с более высокой эффективностью, поскольку собственная ДНК фага защищена метилированием также, как и ДНК хозяина - Е. coli. Однако, если фаг пытается инфицировать другой штамм Е. coli, содержащий другой фермент рестрикции, последний будет быстро атаковать ДНК фага. Насколько успешным окажется инфицирование в этом случае - определит «соперничество» между системой метилирования клетки и ферментом рестрикции. Степень защиты от инфекции очень высока, а ДНК фага А достаточно длинна и может включать несколько участков, на которые способны воздействовать ферменты рестрикции.
Когда неметилированная в этих участках ДНК фага входит в клетку, для успешного ее инфицирования процесс метилирования должен выиграть все «раунды соревнования» у фермента рестрикции: единственный разрез в ДНК фага будет сопровождаться потерей его способности к репродукции в клетке.
Теперь обратимся к технологии выделения генов.
Клонирование гена, или как выделяют гены
В этом разделе мы опишем два метода, используемые при получении рекомбинантной ДНК. Первый из них основан на выделении гена из ДНК.
Возьмем в качестве примера ДНК человека, хотя методы применимы к ДНК из любого источника. Клоны генов представляют собой фрагменты ДНК, идентичные по последовательности оснований участку клеточной ДНК, содержащему нужный ген. Второй метод, заключается в выделении кДНК человека («к» обозначает комплементарный). кДНК представляет собой двухцепочечную копию мРНК. Геномная ДНК и кДНК отличаются друг от друга тем, что первая содержит интроны, а вторая - нет. Это различие имеет важное практическое значение. Дело в том, что клетки бактерий (подобных Е. coli) часто используют для получения инсулина или гормона роста человека. Поскольку первичный РНК-транскрипт генов Е. coli не подвергается сплайсингу (см. с. 271), клонированные гены эукариот (содержащие интроны) не могут управлять синтезом белка в этих клетках. В то же время Е. coli транскрибирует кДНК в мРНК-подобный транскрипт (при условии, что соответствующие транскрипционные сигналы помещены на эту кДНК). Транскрипты будут управлять синтезом белка в Е. coli; для этого необходимо также, чтобы с кДНК были внесены соответствующие трансляционные сигналы. Иногда выделение искомого гена начинают с получения соответствующей этому гену кДНК, которая затем может быть использована в качестве гибридизационного зонда (см. ниже). Такой подход оправдан тогда, когда можно получить препараты, обогащенные одним видом мРНК, например, индуцибельного фермента. Такой препарат мРНК можно использовать в качестве гибридизационного зонда для выделения кДНК.
Таким образом, клоны генов нужны для изучения структуры гена, а клоны кДНК используются при наработкке инсулина человека или других белков в клетках Е. coli.
Мы выбрали два метода - геномное клонирование и клонирование кДНК, потому что они хорошо иллюстрируют общие принципы клонирования гена.
Что такое клонирование?
Клоном называют множество идентичных копий, образованных из одного предшественника. Клонирование гена или кДНК позволяет получить большое число копий фрагмента ДНК.
Сущность клонирования заключается во введении участка ДНК, полученного, например, в результате рестрикции общей ДНК, в бактериальную клетку, где он будет реплицироваться. Одиночная клетка даст начало колонии, содержащей большое число идентичных клеток: например, при выращивании на агаре в течение ночи одиночная клетка Е. coli произведет ~107 клеток в одной колонии. Кроме того, в каждой клетке может существовать множество копий вектора, содержащих свой фрагмент встроенной ДНК. Поскольку это произойдет с каждым фрагментом ДНК рестрикционной смеси, клонирование обеспечивает как амплификацию (увеличение числа копий), так и разделение каждого фрагмента ДНК. Последнее объясняется тем, что клетки Е. coli, содержащие различные векторы, легко отделить друг от друга, выращивая их в колониях на питательных чашках. Если вам удастся выбрать из тысяч различных колоний ту, которая содержит интересующий вас фрагмент ДНК, вы сможете получить фактически неограниченное число копий такого фрагмента, выращивая чистую культуру выбранной колонии.
Выделение клона гена человека
Создание библиотеки генов человека
Прежде всего нужно выбрать клонирующий вектор, который может принять фрагмент ДНК человека, а затем реплицироваться в клетках Е. coli. Для этого есть несколько возможностей, и мы выберем в качестве вектора фаг λ (см. с. 314). Центральная часть молекулы ДНК фага может быть заменена фрагментом ДНК человека без ухудшения возможности репликации такого рекомбинантного фага в клетках Е. coli. Важные для собственной репликации гены фага будут располагаться по обе стороны от встроенного фрагмента. Фаг λ может принять фрагмент чужеродной ДНК длиной 15-20 тысяч пар нуклеотидов (т. п. н.) без ущерба для его репликации. Такой размер фрагмента удобен для большинства случаев геномного клонирования.
Как конструируются рекомбинантные молекулы?
В основе этого метода лежит использование «липких концов» ДНК. Многие ферменты рестрикции, например, ЕсоRI, осуществляют не прямой разрез через обе цепи двухцепочечной ДНК, а «ступенчатый».
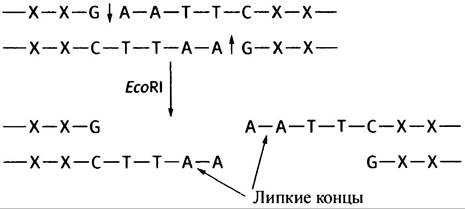
Это приводит к образованию липких концов, причем «выступающие» комплементарные последовательности могут автоматически спариваться друг с другом (в данном случае - А с Т). Если смешать два фрагмента ДНК, имеющих идентичные липкие концы, они соединятся (рис. 24.1). Если же смешать предварительно подвергнутые рестрикции ДНК человека и фага X, липкие концы будут способствовать образованию рекомбинантных молекул ДНК фага, как описано выше. Для ковалентного связывания разрывов используется фермент лигаза, катализирующий образование связи между 3'-ОН- и 5'-фосфорильной группой.
Рис. 24.1. Основные этапы получения рекомбинантных ДНК, используемые при создании библиотеки ДНК на основе фага λ. Следует отметить, что благодаря липким концам могут иметь место другие виды ассоциации фрагментов ДНК; но в головку фага будут упаковываться только те рекомбинантные молекулы, в центре которых расположен фрагмент ДНК человека (15-20 т. п. н.), а по бокам - фрагменты фаговой ДНК
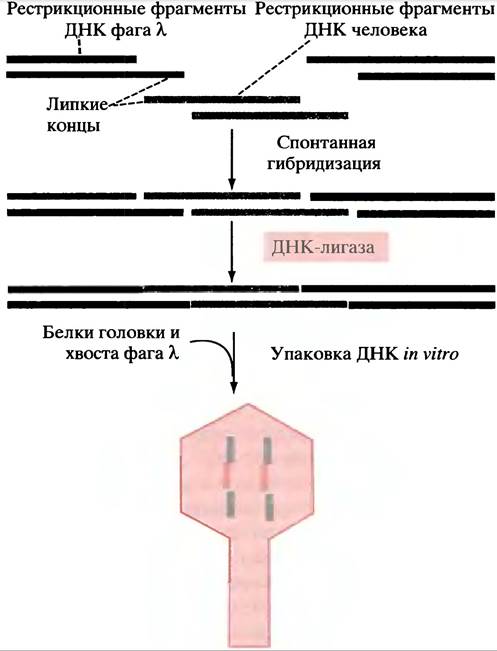
При добавлении рекомбинантных молекул к белковым компонентам фага λ происходит самосборка фага, и рекомбинантная ДНК автоматически пакуется в его головку с образованием зрелого фага.
Упаковка рекомбинантной ДНК в инфекционные фаговые частицы произойдет только в том случае, если фрагмент ДНК человека не более 15-20 т. п. н. будет иметь на своих концах ДНК фага λ. Таким образом проходит отбор рекомбинантных молекул (при получении фрагментов ДНК человека предпринимаются определенные меры, чтобы их размеры соответствовали требованиям, но мы их рассматривать не будем). Фаг λ несущий молекулу рекомбинантной ДНК, используется для заражения клеток Е. coli посредством процедуры, в ходе которой инфицируется лишь небольшая порция клеток. Это необходимо для увеличения вероятности проникновения одной фаговой частицы в данную клетку. Важно также, чтобы штамм Е. coli был мутантным по гену фермента рестрикции. В конечном итоге полностью фрагментированный геном человека будет находиться в культуре клеток Е. coli (по одному фрагменту на клетку). Такое «полное собрание» генома человека, распределенное по «отдельным томам» рекомбинантных молекул фаговых частиц, заключенных в «суперобложку» бактериальной клетки, называют геномной библиотекой. Культура клеток затем наносится на твердую питательную среду и инкубируется. Неинфицированные клетки растут, образуя непрозрачный «газон» по всей поверхности чашки. Среди них находятся и инфицированные фагом клетки, колонии которых проявляются в виде пятна, или бляшки. Происхождение последней обусловлено тем, что после размножения фага в клетке происходит ее лизис, и фаговые частицы инфицируют соседние клетки, разрушая «газон», образованный исходно неинфицированными бактериями. Каждая бляшка содержит множественные копии фага, размножившиеся из одиночной инфекционной частицы, и несет одну и ту же копию фрагмента ДНК человека (рис. 24.2).
Если мы сможем идентифицировать бляшку, которая содержит фаг, несущий требуемый ген, то это позволит вырастить неограниченное число фага в Е. coli и выделить его ДНК. Как же идентифицировать нужную бляшку?
Скрининг бляшек на выбранный ген человека
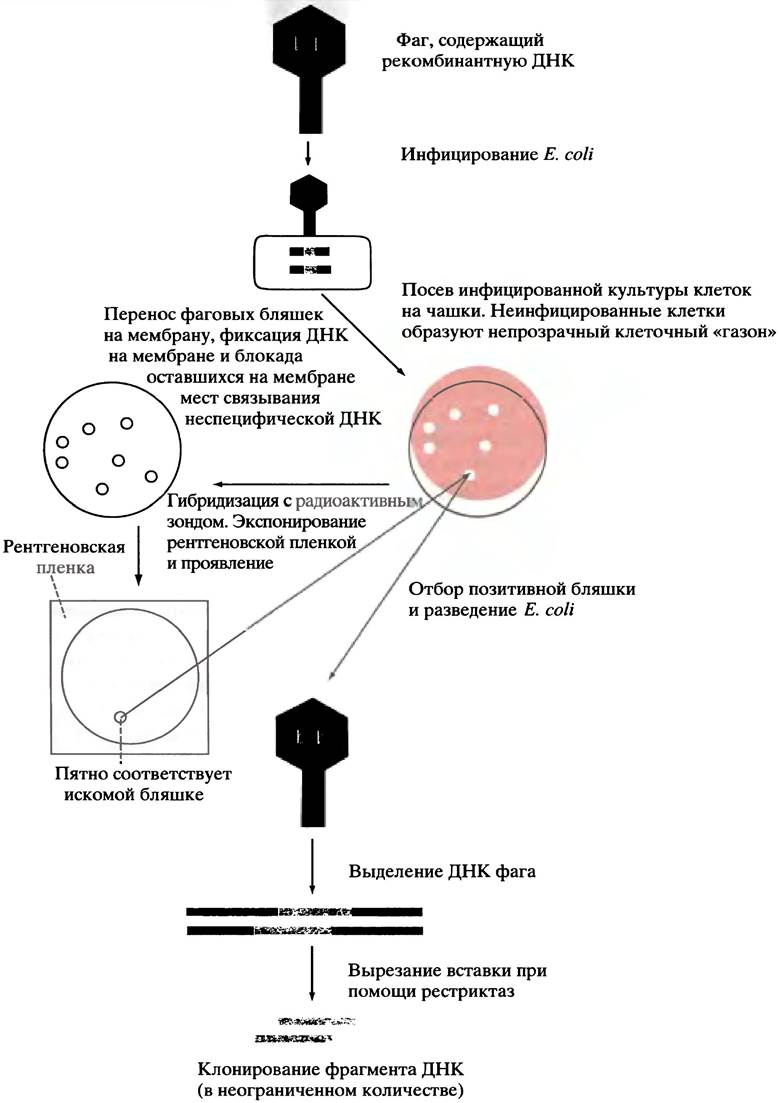
Для идентификации фага, несущего искомый ген человека, наиболее широко используется ДНК-ДНК-гибридизация. Для этого необходим зонд гибридизации, состоящий из короткого фрагмента ДНК (длиной около 20 нуклеотидов), последовательность оснований которого комплементарна известной последовательности в гене. Метод предусматривает перенос фага с каждой бляшки на мембрану с ДНК-адсорбентом. Если наложить мембранный диск на поверхность культивируемой чашки (не забывая его пометить, чтобы потом можно было идентифицировать положение каждой бляшки), то фаг с каждой бляшки прилипнет к мембране. Затем мембрану обрабатывают щелочью для разрушения комплекса белок-ДНК (головки фага), высвобождения и разделения цепей ДНК. Одноцепочечная ДНК фиксируется на мембране, которую затем обрабатывают неспецифической ДНК, чтобы предотвратить фиксацию на мембране зонда. Для гибридизации с любой комплементарной ДНК гибридизационный зонд инкубируется с мембраной (предварительно, с помощью ферментативного фосфорилирования с использованием меченого АТР, в него вводится радиоактивная метка). При этом к зонду «прилипает» только комплементарная ДНК - ген-мишень. Затем при помощи рентгеновской пленки определяют положение гибридизованного зонда (см. рис. 24.2). Идентифицировав бляшку, содержащую нужный ген, можно вырастить неограниченное количество фага с этой бляшки, инфицируя Е. coli, а после этого извлечь из ДНК фага вставки ДНК человека, используя соответствующую рестриктазу.
Рис. 24.2. Этапы клонирования гена с использованием бактериофага λ в качестве вектора
Как получить соответствующий зонд - с нуклеотидной последовательностью, комплементарной последовательности гена, если структура последнего неизвестна? В этом случае можно работать в обратном направлении, опираясь на генетический код (см. табл. 22.1). Если полностью или частично известна последовательность аминокислот в белке, кодируемом интересующим нас геном, мы можем сделать вывод о последовательности оснований в этом гене. Конечно, из-за вырожденности генетического кода такой обратный подход не всегда приемлем. Там, где аминокислота имеет несколько кодонов, мы не можем дедуктивно установить, какой из
них действительно использовался клеткой для синтеза белка (ученым, клонирующим гены, доставляет удовольствие найти в секвенируемых фрагментах метионин или триптофан, кодируемые только одним триплетом). В тех случаях, когда необходимо учитывать вырожденность генетического кода, используют методы, предусматривающие синтез смеси зондов. Специальные приборы легко синтезируют короткие зонды ДНК с определенной последовательностью оснований. Зонд завершает добавленная ферментативным способом радиоактивная фосфатная группа. Если такой зонд недоступен, используют альтернативные возможности скрининга, которые в этой книге не рассматриваются.
Рассмотрим выделение клона кДНК.
Клонирование кДНК человека
Создание библиотеки кДНК человека
Принцип метода заключается в том, что из клеток человека, в которых экспрессируется интересующий нас ген, выделяется мРНК; затем она копируется in vitro в двуцепочечную ДНК, а последняя клонируется. Значительное преимущество этого подхода состоит в выборе ткани, в которой искомый ген высоко активен, следовательно, соответствующая мРНК находится в изобилии.
Среди всей РНК клетки доля мРНК невелика, однако в клетках человека все мРНК (за редким исключением) имеют поли-А-«хвосты» (см. с. 270). Если препарат РНК пропустить через колонку с инертной матрицей, несущей синтетический, лиганд олиго-dT (короткая одноцепочечная «ДНК», составленная только из оснований Т), рибосомная и транспортная РНК проходят через колонку не задерживаясь, а мРНК связывается с ней за счет образования водородных связей А = Т между хвостом и лигандом. Затем мРНК можно элюировать, используя растворы низкой ионной силы.
Изолированная смесь мРНК копируется in vitro в ДНК с использованием полимеразного домена обратной транскриптазы вируса (см. с. 313). Образуется дуплекс РНК-ДНК. РНК разрушается при обработке NaOH или РНКазой, а одноцепочечная ДНК превращается в двуцепочечную под действием свободной от экзонуклеазы ДНК-полимеразы I, выделенной из бактериофага.
Для клонирования молекул кДНК в качестве клонирующего вектора можно использовать фаг к, как это уже было описано для геномных библиотек. Молекулы кДНК не имеют липких концов, однако есть методы ферментативного присоединения липких концов к так называемым молекулам с тупыми концами. Используются и другие методы лигирования «тупых концов».
Что можно сделать с клонированной ДНК?
Итак, мы имеем фаг λ, содержащий нужную геномную вставку (ее называют также инсертом) ДНК или инсерт кДНК. Мы можем получить его в любых количествах,
инфицируя клетки Е. coli, затем выделить ДНК и вырезать инсерты путем рестрикции.
Дальнейшее использование последних для различных целей почти всегда включает встраивание интересующей нас ДНК в другой клонирующий вектор - наиболее часто в бактериальную плазмиду. Фаг к удобен для создания библиотек, поскольку он может принять большие фрагменты ДНК (около 15 т. п. н.) и эффективно инфицировать клетки, что позволяет получить полную геномную библиотеку или комплексную библиотеку кДНК. Однако, когда дело доходит до изучения клонированного фрагмента геномной ДНК или кДНК, недостатком фага становится его собственная большая ДНК по обе стороны от инсерта (около 30 т. п. н.), которая необходима для его репликации, но не представляет никакого интереса для решения поставленных задач. Плазмиды намного меньше и более удобны в обращении. Они не могут принять больших фрагментов чужеродной ДНК подобно фагу к, но если, например, цель работы заключается в секвенировании гена, выделенного с помощью фага к, то этот ген разрезается на фрагменты, которые потом встраивают в плазмиды (см. ниже), и каждый фрагмент секвенируют. Аналогичным образом можно обработать клоны кДНК. Они достаточно малы для встраивания в экспрессирующие векторы, в том числе плазмиды Е. coli, но требуют добавления соответствующих транскрипционных и трансляционных сигналов. При инфицировании ими клеток Е. coli белок, кодируемый кДНК, может быть получен в нужных количествах. Теперь мы рассмотрим бактериальные плазмиды.
Бактериальные плазмиды
Клетка Е. coli имеет одну главную кольцевую хромосому, несущую тысячи генов, которые составляют большую часть генетического материала клетки. Однако в цитоплазме есть также и отдельные мелкие минихромосомы, или плазмиды: кольцевые молекулы ДНК, несущие небольшое количество генов и играющие защитную роль в клетке. Обычно они несут гены, придающие клетке устойчивость к антибиотикам, кодируя, например, разрушающий их фермент. Каждая плазмида способна к репликации, что обеспечивает ее удвоение в клетке.
Плазмиды неинфекционны в том смысле, в каком инфекционен бактериофаг. Однако, если клетки Е. coli обработать, например, СаСl2 при 0° С, а затем повысить температуру до 42° С то они становятся компетентными - способными к принятию плазмиды.
Предположим, что мы имеем дело с клонированной молекулой кДНК, выделенной по вышеописанному методу. Сначала выбранная нами плазмида разрезается
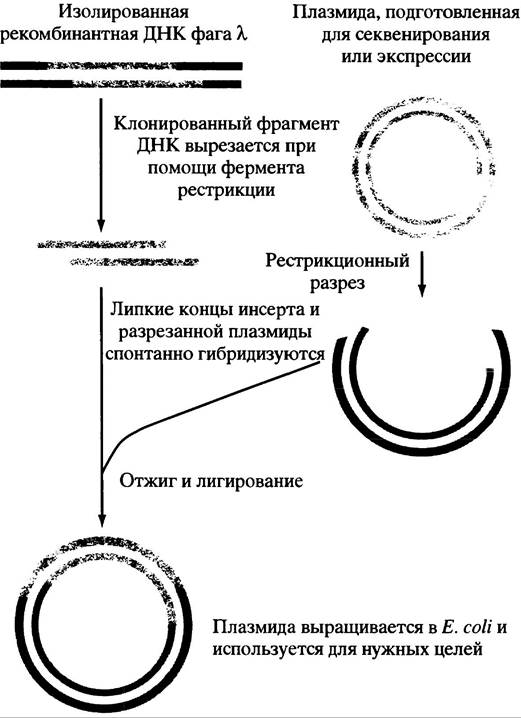
ферментом рестрикции, а к молекулам кДНК добавляются соответствующие липкие концы (мы не будем описывать, как это делается). Когда разрезанные плазмиды и модифицированная кДНК смешиваются и лигируются (т. е. их концы соединяются ферментативным путем) - образуются рекомбинантные плазмиды (рис. 24.3). Во избежание излишнего усложнения рассматриваемого процесса мы не описываем здесь меры, предотвращающие восстановление исходных кольцевых плазмид и способствующие образованию рекомбинантных плазмид. Клетки Е. coli инфицируют рекомбинантными плазмидами и выращивают на агаровых чашках таким образом, что каждая колония вырастает из одной клетки. В результате плазмидного заражения инфицируется только небольшая часть клеток, и возможность захвата двух плазмид одной клеткой минимальна. Поскольку нетрансформированные клетки численно намного превосходят трансформированные, нужен быстрый метод отбора последних.
Рис. 24.3. Перенос инсерта (вставки) ДНК из фага λ в плазмиду. Фрагмент ДНК, который клонировали в фаге или в предназначенной для экспрессии кДНК, с целью наработки нужного белка переносят в Е. coli в составе специально сконструированной плазмиды
Одна из сконструированных плазмид, используемых для клонирования, - pBR322; она несет встроенные в ее ДНК гены устойчивости к ампициллину и тетрациклину. Каждый из этих генов имеет различный участок рестрикции. Предположим, что чужеродный фрагмент ДНК встроен в ген тетрациклина; последний уже не может управлять синтезом белка, придающего устойчивость к этому антибиотику. Клетка, несущая такую плазмиду, будет устойчива к ампициллину, но чувствительна к тетрациклину. Клетка, содержащая плазмиду без вставки ДНК, будет устойчива к обоим антибиотикам, в то время как клетка, не содержащая плазмиду, будет чувствительна к ним. Это служит основой для отбора только тех клеток, которые содержат интересующую вас плазмиду.
Определение последовательности оснований клонированного фрагмента ДНК
Вся информация в гене заключена в последовательности его оснований, определение которой имеет первостепенное значение. Существует два метода секвенирования ДНК(так называется этот процесс). Один из них - прямой химический метод Максама-Гилберта. Другой, используемый наиболее часто, основывается на репликации ДНК и называется методом Сэнгера, или дидезокси-методом.
Схема дидезокси-метода секвенирования ДНК
Предположим, что у вас есть клонированный фрагмент ДНК, который вы хотите секвенировать. Он встраивается в специально сконструированную плазмиду, размножающуюся вместе с инфицированной ею клеткой Е. coli.
Плазмиды выделены, так что у вас в распоряжении имеется большое число рекомбинантных плазмид, содержащих выбранный фрагмент ДНК. Процедура секвенирования (см. ниже) требует, чтобы предназначенный для секвенирования фрагмент копировался in vitro ДНК-полимеразой. Помимо 4 дезоксинуклеозидтрифос- фатов (dNTPs) для этого необходимо: 1) чтобы копируемая ДНК была одноцепочечной; 2) чтобы праймер гибридизовался со стартовым участком (напомним, что ДНК-полимераза не может инициировать новую цепь). Предназначенные для секвенирования плазмиды конструируют таким образом, что у 3'-конца встроенного клонированного фрагмента всегда располагается одна и та же нуклеотидная последовательность (плазмидная ДНК). Зная ее, можно синтезировать комплементарный (олиго)нуклеотид, который и будет служить праймером.
Инкубация предназначенной для секвенирования одноцепочечной ДНК с праймером, полимеразой I (свободной от экзонуклеазы), dАТР, dGТР, dСТР и dTTР обеспечивает копирование фрагмента ДНК. Один из трифосфатов (или праймер) должен быть радиоактивным, так что все новые ДНК оказываются мечеными.
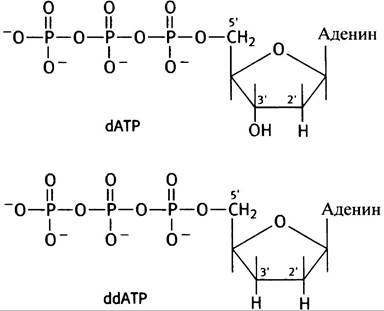
При проведении секвенирования решающими являются два момента. Первый связан с использованием электрофоретического метода разделения молекул ДНК, которые в электрическом поле двигаются вдоль пластины акриламидного геля со скоростью, определяемой длиной цепи: чем короче цепь ДНК, тем быстрее она движется. Каждое присоединение нуклеотида изменяет подвижность цепи. На геле цепям с различной подвижностью соответствуют полосы, причем эти полосы соответствуют цепям, которые отличаются друг от друга на один нуклеотид. Их идентифицируют при помощи радиоавтографии, которая включает экспонирование геля с рентгеновской пленкой, чувствительной к радиоактивности. Второй момент касается дидезокси-производных нуклеозидтрифосфатов (ddNТРs). ДНК-полимераза добавляет 3'-нуклеозид к 3'-ОН-концу растущей цепи ДНК. У дидезоксиNТРs 3'-ОН-группа отсутствует (рис. 24.4); они могут быть добавлены к цепи посредством их 5'-Р-группы, но после этого цепь наращиваться не может.
Рис. 24.4. Структуры дезоксиАТР dАТР) и дидезоксиАТР (ddАТР). Отсутствие 3’-ОН-группы означает, что в момент присоединения ddNТР к растущей цепи последняя обрывается
Важно подчеркнуть, что, когда мы говорим о секвенируемом «фрагменте» ДНК, в эксперименте участвуют множество копий этого «фрагмента». Даже ничтожное количество ДНК содержит большое число молекул (помните, что 1 моль любого химического вещества содержит астрономическое число 6,03-1023 молекул). Предположим, что для копирования мы располагаем всеми четырьмя dNTPs и небольшим количеством одного дидезоксиNТР; для примера возьмем дидезоксиАТР (А). Количества завершенных цепей достаточно для того, чтобы с помощью электрофореза выявить их на геле в виде отдельных полос. Рост цепей будет продолжаться до тех пор, пока к ним не будет добавлен А; отсутствие 3'-ОН-группы делает невозможным присоединение следующего нуклеотида.
Как все это интерпретировать в виде последовательности оснований?
Предположим, что секвенируемый фрагмент ДНК имеет показанную ниже последовательность, которая включает основания Т:
![]()
Если копирование происходит в присутствии дидезоксиАТР (вместе с избытком дезоксиАТР), то при добавлении каждого А рост части цепей будет заканчиваться, и в конечном итоге образуются следующие цепи (связанные с праймером).
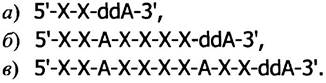
На геле (называемом секвенирующим) эти цепи будут видны в виде полос (рис. 24.5, столбик слева).
Рис. 24.5. Радиоавтография секвенирующего геля. Последовательность полинуклеотида читается снизувверх. а, б, в - Полосы, соответствующие полинуклеотидам, образованным в присутствии дидезоксиАТР
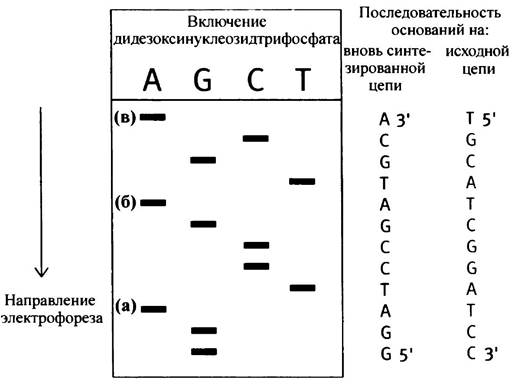
Если вторая инкубационная смесь содержит дидезоксиТТР вместо дидезоксиАТР, то образуется набор цепей, оканчивающихся на Т. Если же в среде присутствуют дидезоксиСТР или GТР, цепи будут оканчиваться на С или в соответственно. При проведении всех четырех инкубаций продукты реакций располагаются на геле друг за другом в виде так называемой секвенирующей лестницы (см. рис. 24.5). С ее помощью легко определить последовательность оснований фрагмента ДНК, которая читается с нижней части геля вверх: это последовательность цепи-копии (т. е. партнера по отношению к матрице), а не матрицы. В результате последовательность располагается в направлении 5' —> 3', поскольку именно в этом направлении всегда происходит синтез.
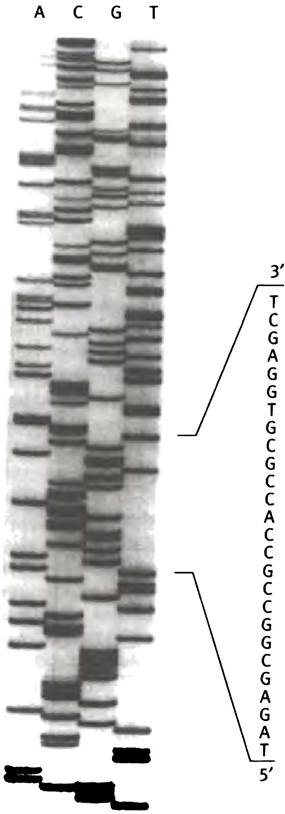
В каждой секвенирующей лестнице можно определить последовательность около 200-300 оснований. Если при использовании различных ферментов рестрикции получаются перекрывающиеся фрагменты исходной кДНК, можно определить ее полную последовательность. Фотография полученной в ходе эксперимента секвенирующей лестницы приведена на рис. 24.6.
Рис. 24.6. Фотография секвенирующей лестницы. А, С, G и Т обозначают дидезоксинуклеозидтрифосфаты, присутствовавшие в инкубационной смеси. Любезно предоставленаDr. Cris Hahn, Departament of Biochemistry, University of Adelaide
Если же кДНК секвенируется с целью определения аминокислотной последовательности белка, то, используя генетический код, определяют правильную рамку считывания (из 6 возможных на 2 цепях).
Описанная выше методика применяется во многих лабораториях. Однако технология секвенирования продвинулась вперед настолько, что позволила сделать этот процесс автоматизированным. Принцип нового метода не отличается от использованного Сэнгером, но предусматривает применение флуоресцентно-меченых нуклеозидтрифосфатов, причем каждый из 4 типов имеет свой максимум флуоресценции. Для инкубации ДНК используется среда, в которой присутствуют все 4 дидезокси- соединения и, конечно, 4 дезоксинуклеозидтрифосфата; продукты анализируются на одной электрофоретической дорожке. Гель автоматически сканируется, и компьютер печатает последовательность оснований.
Использование полимеразной цепной реакции (ПЦР) для амплификации сегментов ДНК
Полимеразная цепная реакция (ПЦР) становится в последние годы одним из наиболее популярных методов работы с ДНК. Она позволяет амплифицировать сегмент молекулы ДНК путем синтеза огромного числа его копий. Для проведения ПЦР достаточно ничтожно малых количеств ДНК (всего из нескольких клеток). Принцип метода прост и изящен. Для проведения ПЦР необходимо знать нуклеотидную последовательность интересующей нас ДНК и синтезировать два олигонуклеотида, каждый из которых комплементарен участку одной из двух цепей молекулы ДНК, примыкающему к выбранному для амплификации сегменту. Эти олигонуклеотиды будут праймерами при синтезе ДНК in vitro. Цепи дуплекса ДНК разделяют нагреванием и копируют выбранный сегмент с использованием синтетических праймеров (рис. 24.7). В конце каждого цикла синтеза дуплексы ДНК, образованные в процессе копирования, разделяются нагреванием для осуществления следующего раунда праймирования и копирования. Использование термостабильных ДНК-полимераз (из термофильных бактерий) исключает необходимость добавления фермента после каждого цикла копирования и разделения цепей в ходе нагревания.
Рис.24.7. Схематичное изображение, иллюстрирующее основной принцип амплификации участка ДНК в ходе полимеразной цепной реакции (ПЦР) а - В центре - участок, выбранный для амплификации с использованием соответствующих праймеров (—> <—); каждый из них комплементарен 3'-концу участка амплифицированной цепи;
б - нагревание приводит к разделению цепей (с этого момента для простоты схемы показана амплификация только одной цепи, хотя в действительности амплифицируются обе); в - инкубационные смеси содержат 4 (dNTPs, термостабильную ДНК-полимеразу и соответствующие праймеры; г - ДНК реплицируется; д - в ходе нагревания цепи разделяются; е - праймируется новая цепь; ж - новая цепь реплицируется; з - нужный фрагмент отделяется. За 25 циклов репликации, нагревания, праймирования и синтеза выбранный участок может быть амплифицирован в миллионы раз. Обратите внимание, что каждый раз происходит праймирование и копирование

До последнего времени можно было амплифицировать участок ДНК, состоящий не более, чем из 2 т. п. н., однако новейшие технологии позволяют амплифицировать фрагменты, включающие до 35 т. п. н. Полученные таким образом копии ДНК, могут использоваться для самых разных целей. Этот метод важен для судебной медицины и в диагностике генетических аномалий плода in utero (см. ниже). Он используется также и в биохимических исследованиях.
Применение технологии амплификации рекомбинантной ДНК
Как уже отмечалось, главная роль клонирования гена заключается в определении последовательности оснований. Это необходимое условие для изучения функции гена и его регуляции. Последовательность оснований ДНК - это прямое «окно» в эволюционные взаимосвязи организмов. Амплификация рекомбинантных ДНК нашла практическое применение и в ряде других случаев, которые мы здесь вкратце разберем.
Экспрессия гена в клетках бактерий и эукариот
Выделенный клон кДНК или бактериальный ген могут быть встроены в специально сконструированный вектор. Например, в плазмиду, в которую включены соответствующие промоторы бактериальной ДНК и сигналы инициации трансляции. Встроенная в каждую плазмиду молекула кДНК экспрессируется; клетка синтезирует из кДНК мРНК и транслирует ее в белок. Таким образом, встроенные гены покариот или кДНК эукариот (помните, что она не имеет интронов) могут быть эффективно транскрибированы, а соответствующие мРНК транслированы, в результате чего можно получить нужный белок.
При помощи описанных методов бактерии (Е. coli и др.) способны производить неограниченное количество белков человека, применяемых в качестве лекарственных средств. Кроме наработки белков, присутствующих в организме человека в ничтожно малых количествах, использование Е. coli исключает возможность присутствия в белке примесей инфекционных агентов. Так продуцируется человеческий инсулин и некоторые другие белки, например, гормон роста. В качестве вакцины, без риска занесения инфекции, используется иммуногенный белок вируса гепатита В, образующийся в дрожжах. В некоторых случаях Е. coli может продуцировать чужеродный белок в таких количествах, что он преципитирует в виде телец включений и не может принимать нужную конформацию. В ряде случаев лишь in vitro происходит правильное сворачивание белка. Бактерии не могут гликозилировать белки, но, если использовать в качестве экспрессионных хозяев животные клетки, например, клетки насекомых, глико- зилированные белки могут образовываться в больших количествах.
Направленный мутагенез
Это очень мощный метод анализа роли отдельных аминокислотных остатков в белке. Рассмотрим случай, когда у нас есть фермент, для которого известна аминокислотная последовательность, установленная либо в результате прямого секвенирования, либо на основании последовательности оснований его гена или кДНК. Теперь мы хотим исследовать роль боковой цепи отдельной аминокислоты в функционировании данного белка. Если бактериальный ген или кДНК эукариот клонировать в экспрессирующемся векторе, например, плазмиде, клетки Е. coli будут продуцировать белок в количествах, достаточных для исследования. Например, в случае фермента нас может интересовать его каталитическая активность или субстратная специфичность. Направленный мутагенез позволяет осуществить специфическое замещение одной или нескольких выбранных аминокислот в белке. Это можно сделать, замещая соответствующий фрагмент выделенного гена синтетическим олигонуклеотидом, последовательность оснований которого такова, что кодирует измененную аминокислоту. Получив мутантный белок, можно затем определить его каталитическую активность.
Трансгенез
Трансгенезом называется встраивание генов животным или растениям. Встраивание нормальных генов пациентам для коррекции дефектных генов (генотерапия) может быть использовано для лечения генетических заболеваний. В качестве векторов для введения генов в хромосомы человека можно было бы использовать ретровирусы, «покалеченные» генетически для предупреждения их репликации. Однако этот способ не позволяет определить инсерционную точку, а беспорядочная инсерция потенциально опасна. В настоящее время разрабатываются методы инсерции гена в специфических точках. Инсерция гена аденозиндезаминазы человека в лейкоциты больных лечит иммунодефицит у детей (см. рис. 18.6). Для инсерции чужеродных генов в хромосомы растений в качестве клонирующего вектора используются природные (но соответствующим образом модифицированные) Ti-плазмиды (от англ. tumor-inducing - опухоль-индуцирующая), содержащиеся в патогенной почвенной бактерии Agrobacteriumtumefaciens. Метод дробовика предусматривает «стрельбу» генами по клеткам растений при помощи специального «ружья». Таким способом конструируются растения, устойчивые к гербицидам; это позволяет уничтожать гербицидами сорняки, не влияя на рост. Трансгенные животные могут также использоваться в качестве «экспрессирующихся векторов». Например, белок человека может нарабатываться после присоединения его гена к сигнальной последовательности гена, ответственного за синтез белка молока, что в конечном итоге приводит к секреции такого белка вместе с молоком. Этот подход был использован для получения белка человека в молоке овец.
Определение генетических аномалий анализом рестрикции или блоттингом по Саузерну
Когда известна структура генов, можно сделать гибридизационные зонды для определения их аномалий. Для этого анализа требуется минимум ДНК, что позволяет проводить пренатальную диагностику плода (при использовании полимеразной цепной реакции для таких анализов достаточно ничтожно малых количеств ДНК). Наличие информации о потенциальных заболеваниях позволяет принять решение о возможном прерывании беременности. Например, при мышечной дистрофии аномалия гена обычно обусловлена делецией части кодирующей ДНК, и это можно определить Саузерн-блот-анализом. Этот анализ, названный по имени автора, включает разрезание ДНК одним или несколькими ферментами рестрикции, разделение фрагментов при помощи гель-электрофореза, перенос разделенных фрагментов на мембрану и зондирование мембраны ген-специфичным радиоактивным зондом гибридизации. Все это позволяет выявить участки ДНК в виде полос. Аномалии гена могут давать различные картины полос, потому что в результате мутации могут появляться новые участки для атаки фермента рестрикции либо разрушаться или удаляться ранее существовавшие. Даже если на геле будет присутствовать много различных фрагментов ДНК, выявляться будут лишь те немногие из них, что гибридизуются со специфическим зондом.
Если локализация гена, обусловливающего возникновение генетической болезни, точно не известна, или он не выделен, можно применить RFLP-анализ - анализ полиморфизма длины рестрикционных фрагментов (англ. restriction fragment length polymorphism). Метод здесь не обсуждается. Его можно использовать для идентификации родственников больного - носителей аномального гена.
Понять принцип применения RFLP-анализа легче всего на примере метода «отпечатков пальцев» ДНК, или фингерпринтинга (англ. fingerprinting) ДНК. Повторяющиеся последовательности в ДНК, разбросанные по всему геному, свидетельствуют о большом полиморфизме сайтов рестрикции: рестрикционные образцы уникальны у каждого человека. Этот метод до сих пор используется в судебной медицине, однако новая технология, базирующаяся на ПЦР-реакции, постепенно его вытесняет. В геноме человека идентифицировано большое число повторяющихся последовательностей. Число повторов в последовательности сильно варьирует у разных индивидуумов. Этот метод заключается в выборе праймерных сайтов с каждой стороны повторяющейся последовательности и ее амплификации при помощи ПЦР.
Подвижность анализируемых образцов ДНК при электрофорезе зависит от числа повторов в амплифицированном сегменте. Выбирая число таких локусов для амплификации, получают картину полос, характерную для каждого индивидуума.
Вопросы к главе 24
1. Чем фермент рестрикции отличается от панкреатической ДНКазы?
2. Фермент рестрикции Е. coli RI разрезает гексамерную последовательность оснований. Такая последовательность должна многократно встречаться в ДНК Е. coli. Почему же фермент не разрушает свою собственную ДНК?
3. Что обозначает термин «липкие концы» применительно к молекулам ДНК?
4. Что такое клон генов? Что такое клон кДНК? Чем они отличаются у эукариот?
5. Каковы этапы подготовки геномной библиотеки в бактериофаге λ?
6. Каковы этапы выделения определенного клона из геномной библиотеки, помещенной в фаг λ?
7. Что такое дидезоксинуклеозидтрифосфат? Какова его функция в секвенировании ДНК по методу Сэнгера?
8. Кратко изложите суть полимеразной цепной реакции и условия ее проведения.
9. Что такое бактериальный экспрессирующий плазмидный вектор?
10. Что такое рестрикционный анализ?