Основы биохимии - Филиппович Ю. Б. 1999
Углеводы и их обмен
Обмен углеводов
Нередко функцию углеводов в обмене веществ сводят только к энергетическому обеспечению химических реакций. Это далеко не так. Бесспорно, что при распаде (окислении) углеводов в организме идет высвобождение энергии, которая запасается далее в макроэргических связях АТФ, и что АТФ, синтезированная сопряженно с окислением углеводов, поставляет энергию для осуществления химических процессов и для других нужд организма. Однако углеводы выполняют еще одну важнейшую функцию в процессе обмена веществ — они являются источником большого числа органических соединений, которые служат исходными продуктами для биосинтеза липидов, белков и нуклеиновых кислот. В углеводах, образующихся в процессе первичного биосинтеза органического вещества, связывается углерод и запасается энергия.
Распад углеводов. Пути распада полисахаридов и дисахаридов. Полисахариды и олигосахариды распадаются до более простых соединений посредством реакций двух типов: гидролиза и фосфоролиза. Классическим примером распада первого типа является гидролиз крахмала, второго — фосфоролиз гликогена.
Ступенчатый характер гидролиза крахмала рассмотрен ранее. Реакция гидролиза крахмала ускоряется амилазами — специфическими ферментами, относящимися к подклассу гидролаз гликозидов (класс гидролаз). В зависимости от характера фермента разрыв гликозидных связей между остатками а-D-глюкопиранозы в молекуле крахмала и присоединение по месту разрыва элементов воды (Н и ОН-группа) может происходить в различных позициях. Соответственно этому конечными продуктами гидролиза крахмала оказываются либо глюкоза, либо мальтоза, либо олигосахариды. Естественно, что в процессе постепенного укорочения молекулы крахмала в результате гидролитического отщепления моносахаридных, дисахаридных или олигосахаридных звеньев на какой-то ступени распада в качестве промежуточных продуктов возникают декстрины. Наличие и динамику их образования легко установить, прослеживая изменение окраски крахмала с иодом от синей до красно-бурой в процессе ферментативного гидролиза крахмала.
В природе найдено несколько различных амилаз.
Глюокоамилаза, или у-амилаза (а-1,4-глюкан глюкогидролаза), ускоряет реакцию гидролиза 1,4-связей в молекуле крахмала, олигосахаридов и даже дисахаридов (например, мальтозы), последовательно отщепляя остатки глюкозы от невосстанавливающегося (не содержащего свободной альдегидной группы или гликозидного гидроксила) конца молекулы. Механизм ее действия можно представить следующей схемой:
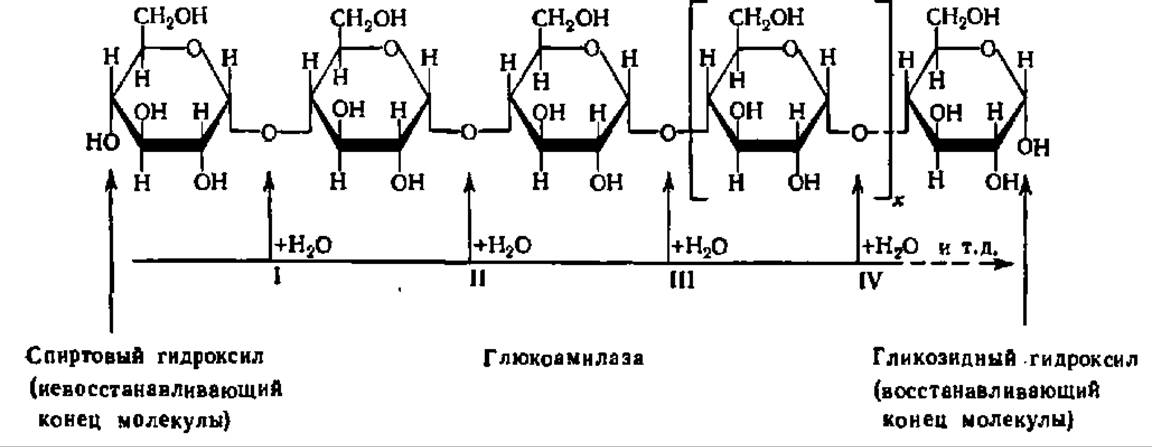
Цифрами I, II, III, IV и т. д. здесь обозначены последовательные реакции гидролиза 1,4-связей в молекуле 1,4-глюкана при каталитическом воздействии глюкоамилазы. Если в молекуле полисахарида есть разветвление, то действие у-амилазы прекращается. Глюкоамилаза ускоряет гидролиз не только крахмала, но и гликогена.
Глюкоамилаза распространена повсеместно; она открыта Е. Л. Розенфельд (1959) в тканях животных, где ярко представлена, как, впрочем, и в плесневых грибах. Из ряда источников глюкоамилаза выделена в гомогенном состоянии. Ее молекулярная масса в большинстве случаев близка к 100000 (у-амилаза из почек человека — 97 000, из печени быка — 107 000, из печени крысы — 114 000, из гриба аспергилла — всего 62 000). Для глюкоамилазы из печени быка доказана мультимерная структура молекулы: 4 субъединицы по 26000 каждая. Обнаружено 2 вида глюкоамилаз — кислая (оптимум pH 4,8—5,0, локализована в лизосомах, КМ для гликогена — 5,45 ∙ 10-3 М) и нейтральная (оптимум pH 6,0—6,5, локализована в микросомальной фракции клетки и в гиалоплазме, КМ для гликогена — 16,25 ∙ 10-3 М). Отсутствие кислой глюкоамилазы у человека связано с тяжелым наследственным заболеванием — гликогенозом; оно состоит в накоплении гликогена в клетках печени, мышц и других органов. Глюкоамилаза иммобилизована и в этом виде применяется в промышленном масштабе для гидролиза крахмала до глюкозы; созданная у нас установка позволяет вести процесс в течение 3 месяцев без заметной потери активности фермента.
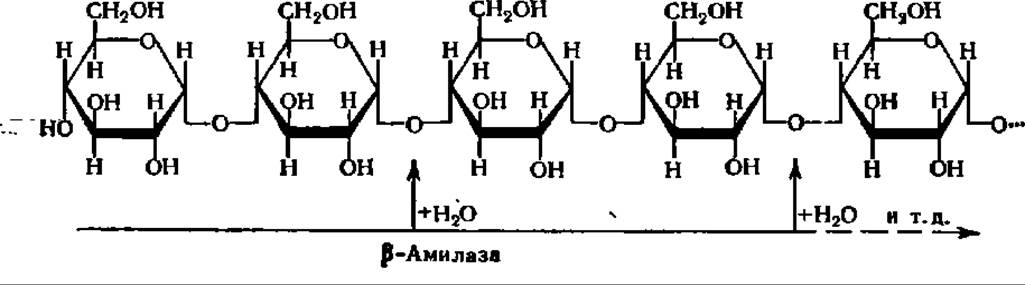
ß-Амилаза (а-1,4-глюкан мальтогидролаза) ускоряет реакцию гидролиза крахмала по 1—4 связям, последовательно отщепляя остатки мальтозы, начиная с нередуцирующего конца молекулы крахмала; ее действие, как и у-амилазы, прекращается в точках разветвления в молекуле крахмала:
Мальтоза, освобождающаяся при гидролизе крахмала под действием ß- амилазы, получается в ß-форме:
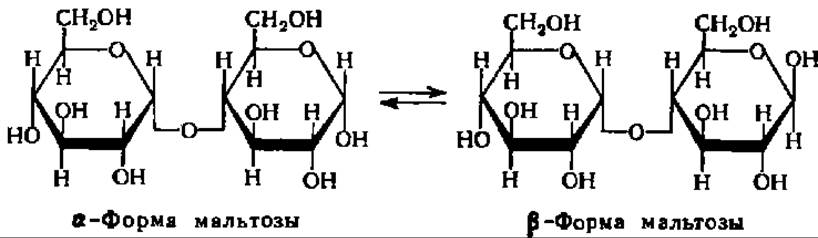
Предполагают, что образование ß-формы мальтозы, т. е. изменение пространственной конфигурации молекулы по месту образующегося гликозидного гидроксила при 1-м углеродном атоме остатка глюкозы, происходит в момент гидролиза а-гликозидной связи. В соответствии с этим фермент и получил свое название. ß-Амилаза характерна только для высших растений, из которых она выделена и получена в виде кристаллов. Молекулярная масса ß-амилазы из батата равна 201000 (4 x 50000).
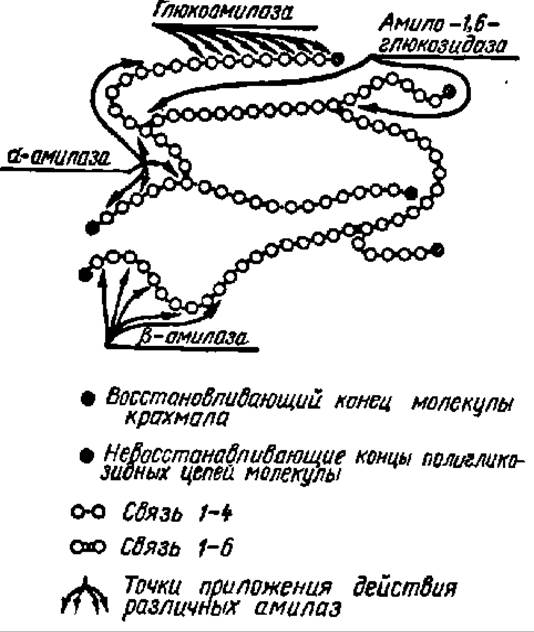
Рис. 105. Схема ферментативного гидролиза крахмала под действием амилаз разных типов (пояснение в тексте)
а-Амилаза (а-1,4-глюкан 4-глюканогидролаза) ускоряет гидролиз 1,4-гликозидных связей в молекуле крахмала без какого-либо определенного порядка, в результате чего сначала возникают олигосахариды, которые тоже подвержены действию ос-амилазы, если они содержат три или более остатков а-D-глюкозы. Таким образом, а-амилаза является эндоамилазой, так как она атакует внутренние гликозидные связи в молекуле крахмала и для ее действия не имеет значения близость или удаленность нередуцирующих концевых остатков полигликозидных фрагментов молекулы крахмала. В качестве главного конечного продукта гидролиза крахмала при участии а-амилазы образуется мальтоза, так как в дисахаридах 1,4-связи под действием а-амилазы не гидролизуются.
Если действие а-амилазы распространяется на спирализованный участок молекулы крахмала или молекулы амилозы, то гидролиз 1,4-гликозидных связей осуществляется латерально, вдоль одного бока спирализованного 1,4-глюкана. Поскольку каждый виток спирали содержит 6—7 остатков а-D-глюкопиранозы, в результате гидролиза возникают гекса- или гептасахариды. Такой механизм действия а-амилазы показан на рис. 103.
Гидролиз крахмала с помощью а-амилазы во всех случаях приводит к образованию а-формы мальтозы (см. с. 316), откуда ведет свое происхождение название фермента. а-Амилаза найдена у всех растительных и животных видов; кроме крахмала она гидролизует также гликоген. Молекулярная масса ос-амилазы из поджелудочной железы равна 45000, из солода — 59000, из мучного хрущака — 68000, из двух видов аспергилла — 51000 и 58000, из бактерий — 96000 (4 x 24000). Выяснена первичная и третичная структура а-амилазы из аспергилла.
Амило-1,6-глюкозидаза ускоряет реакцию гидролиза 1,6-связей в молекуле крахмала (амилопектина), расщепляя, таким образом, молекулу крахмала в точках разветвления полиглюкозидной цепи и образуя олигосахариды с тем или иным числом остатков а-D-глюкозы в них в зависимости от длины отщепляемой цепи. Амило-1,6-глюкозидаза характерна для животных тканей. Однако это не значит, что ее нет в растительном мире: у растений этот же фермент называют R-энзимом, а у микроорганизмов — изоамилазой.
Общая схема гидролиза крахмала при участии перечисленных выше ферментов приведена на рис. 105.
Характерная особенность амилаз — отсутствие абсолютной специфичности действия. При их участии гидролизуются различные соединения: амилоза, амилопектин, гликоген, олигосахариды и родственные им вещества, построенные из остатков a-D-глюкопиранозы и содержащие в молекулах 1,4- и 1,6-связи. По-видимому, все амилазы являются металлопротеинами (содержат Zn2+ и Са2+), некоторые из них мультимеры; предполагают, что ионы металлов способствуют становлению молекулы мультимера из протомеров.
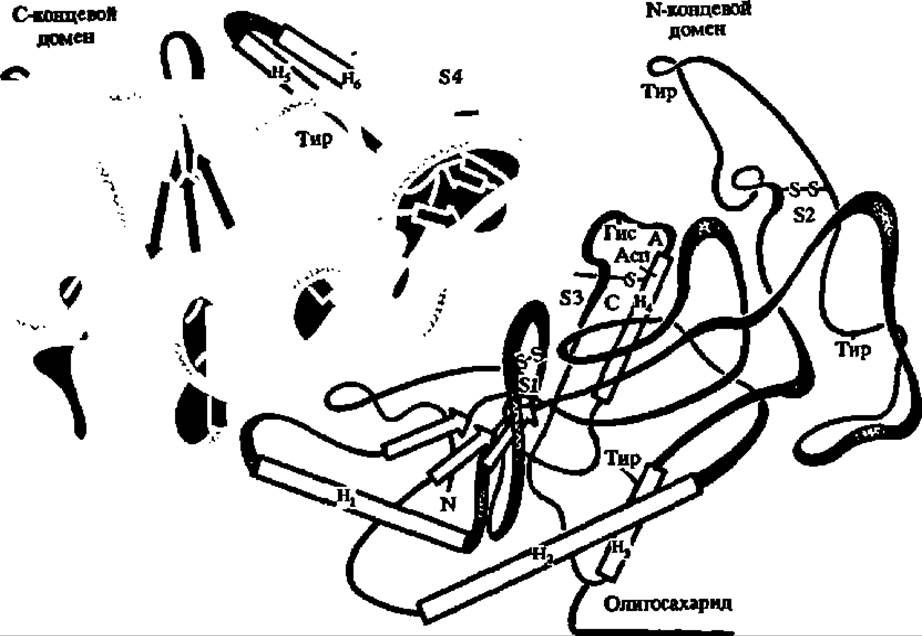
Рис. 106. Третичная структура f-амилазы, продуцируемой Aspergillus orizae (така-амилаза А)
Молекула представляет эллипсоид (8,0 ∙ 4,5 -4,5 нм), содержит 452 аминокислотных остатка, чередование которых выяснено, а также олигосахарид, составленный из 2 остатков глюкозамина и 6 остатков маннозы. Имеет ясно выраженную двухдоменную структуру; на границе доменов располагается активный центр (А), включающий радикалы аспарагиновой кислоты н гистидина, н Са3+-связывающий центр (С); остаток тирозина, нависающий над активным центром, принимает, как и Са2+, участие в связывании субстрата. Третичная структура фиксируется четырьмя —S—S-мостиками (S1—S4).
8а-спиралей обозначены прямоугольниками (Н1—Н8), а 8β-слоев—стрелками. Разрешение — 0,3 нм
Активные центры амилаз образованы радикалами гистидина, аспарагиновой и глутаминовой кислот, а также тирозина (рис. 106). Последнему приписывают функцию связывания субстрата, а первым трем — каталитическую функцию. В соответствии с представлениями о структуре активного центра амилаз предполагают на 1-м этапе следующий механизм распада гликозидных связей при их посредстве:
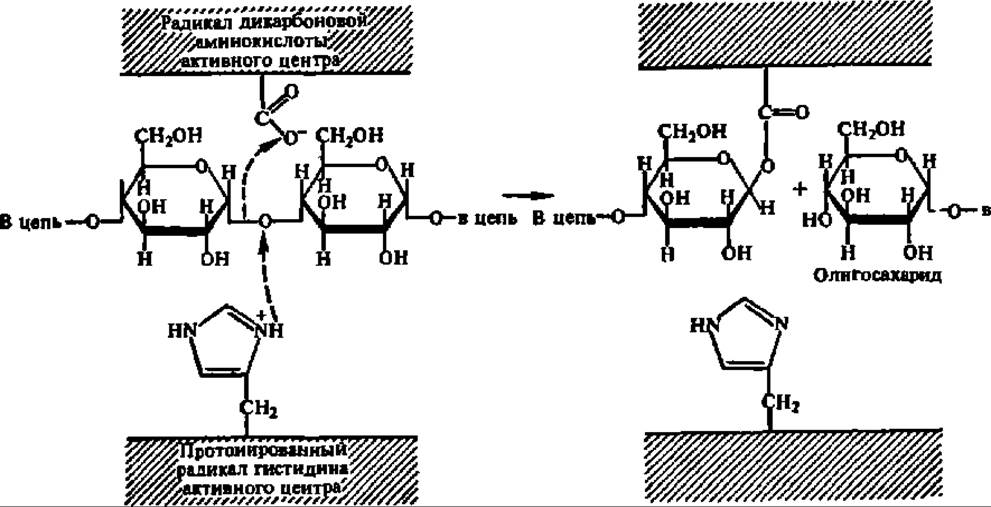
На 2-м этапе в активный центр фермента входит молекула воды, присоединение ОН-группы которой обеспечивает распад возникшей сложноэфирной связи и высвобождение радикала дикарбоновой аминокислоты; протон воды присоединяется к депротонированному на 1-м этапе радикалу гистидина:
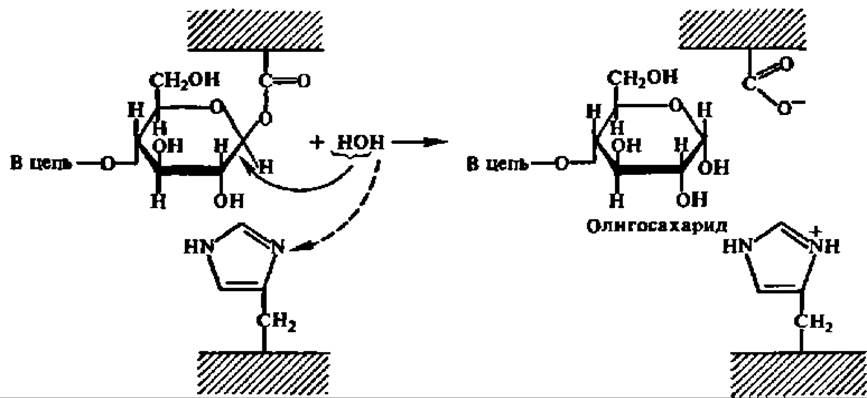
В результате восстанавливается структура активного центра фермента, и он готов к осуществлению следующего каталитического акта в гидролизе гликозидной связи. Есть достаточно оснований считать, что не только амилазы, но и все другие гликозидазы (см. ниже) действуют в соответствии с приведенным выше механизмом.
Особую роль в осуществлении гидролиза гликозидных связей играют имидазольные радикалы гистидина, присутствующие в каталитическом центре амилаз. Именно при их посредстве идет перенос протонов, столь необходимый для обеспечения каталитического акта. Такая функция радикалов гистидина характерна и для каталитических центров ряда других гидролаз (см. с. 229, 263 и 406).
Аналогично крахмалу и гликогену гидролизуются другие природные полисахариды: целлюлоза — при участии целлюлазного комплекса ферментов (состоит из эндо-1,4-β-люканазы, экзоцеллобиогидролазы, целлобиазы и экзо-1,4-β-глюкогидролазы), инулин — с помощью инулиназы (2,1-ß-D-фруктап фруктаногидролазы), хитин — хитиназы, ксилан — ксиланазы, гиалуроновая кислота — гиалуронидазы, пектиновые вещества — комплекса пектинолитических ферментов (пектинэстеразы, полигалактуроназы и др.) и т. п. Во всех случаях полисахариды распадаются до соответствующих моносахаридов или их производных (иногда до дисахаридов), из которых составлены молекулы указанных полисахаридов. Некоторые из гидролаз, перечисленных выше, широко распространены в природе, другие (целлюлаза, инулиназа) — присущи лишь определенным видам растений, животных и микроорганизмов. Особое внимание в последние годы привлекают целлюлазы, хитиназы, 1,3- и 1,6-ß-глюканазы и лихеназы (гидролизуют гетерополисахариды с перемежающимися 1,3- и 1,4-гликозидными связями), так как они деструктируют растительные и микробные остатки, являющиеся экологически опасными отходами производства, позволяя при этом получать глюкозу и N-ацетилглюкозамин. Некоторые из них, например, хитиназы из Bacillus cereus, множественны (68, 52 и 38 кДа), выделены и очищены до гомогенного состояния; они ускоряют реакцию гидролиза хитина при pH = 4 и t = 60° С до хитобиозы, но не до N-ацетил-β-О-глюкозамина.
В свою очередь, дисахариды, возникающие в ряде случаев при гидролизе полисахаридов (мальтоза при гидролизе крахмала, целлобиоза при гидролизе целлюлозы) или существующие в организме в свободном виде (лактоза, сахароза, трегалоза и т. п.), гидролизуются при каталитическом воздействии f- и ß-гликозидаз до индивидуальных моносахаридов. Все гликозидазы, за исключением трегалазы (а, а-трегалоза-глюкогидролазы), отличаются широким спектром специфичности, ускоряя гидролиз практически любых гликозидов, являющихся производными того или иного а- или ß-моносахарида. Так, f-глюкозидаза ускоряет реакцию гидролиза a-глюкозидов, в том числе мальтозы; ß-глюкозидаза — ß-глюкозидов, в том числе целлобиозы; ß-галактозидаза — ß-галактозидов и среди них лактозы и т. д. Примеры действия а и ß-глюкозидаз были приведены ранее.
Гликозидазы, кроме гидролазной активности, как правило, обладают также гликозилтрансферазным действием и ускоряют процессы переноса гликозильных остатков на те или иные субстраты. Так, например, целлобиаза из аспергилла (М = 142 000) при действии на целлобиозу образует не только ß-D-глюкозу, но и трисахариды, т. е. переносит остаток ß-D-глюкозы с одного дисахарида на другой, что особенно ярко выражено в начале ферментативного процесса. Сейчас все более укрепляется мнение, что трансгликозилирование является одним из существенных элементов распада полисахаридов, олигосахаридов и дисахаридов, сопутствующих их гидролизу.
Второй тип распада полисахаридов и олигосахаридов представлен реакциями фосфоролиза. Если попытаться выразить реакцию фосфоролиза в виде суммарного химического уравнения, то она сводится к присоединению элементов фосфорной кислоты (водорода и кислотного остатка) по месту разрыва гликозидной связи между остатками моносахаридов в молекулах олиго- или полисахаридов:
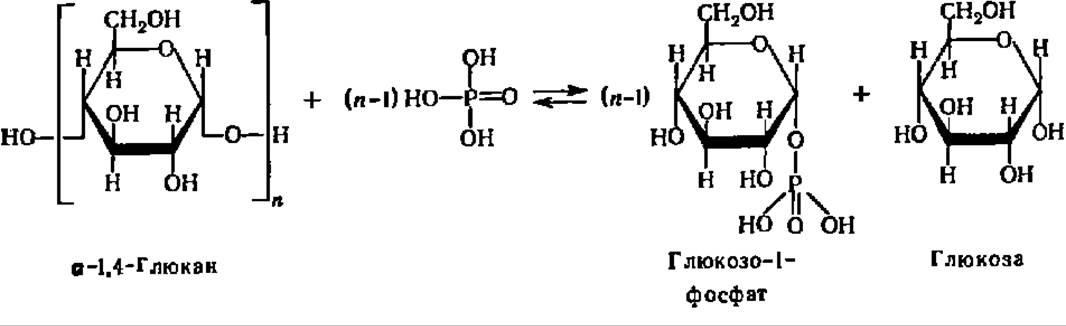
Опираясь на это формальное представление, указанную реакцию расценивали на первых порах как процесс, полностью аналогичный реакции гидролиза, с тем лишь отличием, что роль воды в данной реакции играет Н3РO4. Поэтому она и была названа реакцией фосфоролиза. Эта реакция ускоряется специфическими ферментами — фосфорнлазами, которые относятся к подклассу гликозилтрансфераз и подподклассу гексозилтрансфераз. Они были открыты Я. О. Парнасом, К. Кори и Г. Кори более полувека тому назад.
Фосфорилазы ускоряют процесс переноса гликозильного остатка с невосстанавливающего конца молекулы полисахарида или олигосахарида на неорганический фосфат. Фосфоролитическому расщеплению подвергаются только 1,4-связи (см. уравнение реакции на с. 128 и 334).
Представленный выше процесс многократно повторяется, и ступенчатый распад a-1,4-глюкана сопровождается выделением большого числа молекул глюкозо-1-фосфата. В случае строго линейного полигликозида фосфоролиз идет до конца, в случае разветвленных полисахаридов — останавливается в точках разветвлений полисахаридной цепи.
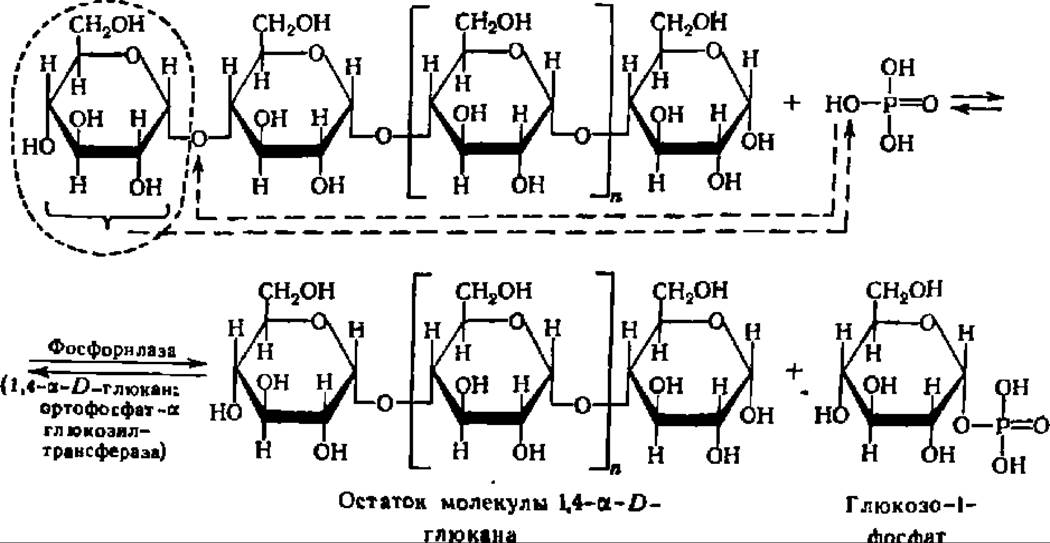
Реакция фосфоролиза полисахаридов широко представлена в природе. Именно так идет распад гликогена, когда он вступает на путь гликогенолиза (см. ниже). Аналогично этому посредством реакции фосфоролиза значительная часть крахмала превращается в глюкозо-1-фосфат при использовании запасов крахмала для нужд растительного организма. Отмечены также случаи фосфоролиза дисахаридов. Так, некоторые бактерии содержат мальтозофосфорилазу, ускоряющую реакцию распада мальтозы на глюкозо-1-фосфат и глюкозу при взаимодействии ее с Н3РО4.
Фосфорилазы — мультимеры высокой молекулярной массы. Наиболее изучена фосфорилаза из мышц кролика. Ее молекула (М = 400 000) состоит из четырех протомеров (4x100000), каждый из которых представлен полипептидной цепью из 841 аминокислотного остатка. Последовательность их расположения установлена; в частности, выяснено, что в положении 14 находится серин, фосфорилированный по ОН-группе радикала, а в положении 679 — лизин, с ε-аминогруппой которого альдиминной связью соединен пиридоксальфосфат, являющийся простетической группой фосфорилазы. Электронно-микроскопически выяснена и третичная структура протомеров, их топография, локализация субстратного и каталитического центров, пространственная компоновка мономеров в тетрамере (рис. 107). Именно в таком состоянии, т. е. в виде тетрамера, гликогенфосфорилаза из мышц кролика проявляет высокую активность в переносе гликозильных остатков на неорганический фосфат. Ее называют фосфорилазой а.
Будучи дефосфорилирована по 14-му остатку фосфосерина каждого из протомеров (это происходит при участии протеинфосфатазы), тетрамерная молекула фосфорилазы а распадается на две равные части (т. е. на два димера), называемые фосфорнлазой b, и частично теряет каталитическую активность. После фосфорилирования протомеров в неполностью активных димерах фосфорилазы b тетрамерная структура восстанавливается и снова идет реакция фосфоролиза. Фосфорилирование димеров осуществляется при участии киназы фосфорилазы b (о киназах см. с. 124), переносящей фосфат с АТФ на радикал остатка серина, находящийся в 14-м положении в полипептидной цепи мономера. Становление тетрамера зависит от присутствия Mg2+, участвующего в образовании ионных связей между протомерами, в том числе, вероятно, за счет фосфатных групп:

Фосфорилаза оказалась ферментом, исследование которого позволило впервые детально изучить вопрос о регуляции активности ферментов за счет реакций фосфорилирования — дефосфорилирования (рис. 108). Дело в том, что действие киназы фосфорилазы b, в свою очередь, тоже регулируется при помощи такого же механизма. Этот фермент имеет огромную молекулярную массу — 1272000 Да. Он построен из 4 субчастиц с М = 318 000, каждая из которых, в свою очередь, составлена из трех протомеров; два из них (А и В) являются регуляторными, а один, С, несет каталитическую функцию, т. е. ускоряет реакцию фосфорилирования фосфорилазы b. Однако протомер С активен лишь в том случае, когда фосфорилирован регуляторный протомер В и дефосфорилирован регуляторный протомер А. Такой способ регуляции активности фермента получил название регуляции за счет фосфорилирования второго регуляторного центра (рис. 109).
Фосфорилирование протомера В, как, впрочем, и протомера А, когда киназа фосфорилазы b инактивируется, в свою очередь, осуществляется при участии киназы, которая является, таким образом, киназой киназы фосфорилазы b. Крайне существенно, что она активна только в присутствии аллостерического регулятора — циклического аденозинмонофосфата (цАМФ), возникающего из АТФ в присутствии аденилатциклазы, деятельность которой контролируется гормонально (см. рис. 108). Естественно, что попеременное дефосфорилирование протомеров А и В при посредстве протеинфосфатазы тоже является условием регуляции активности киназы фосфорилазы b. Таким образом, активность фосфорилаз, как и, следовательно, процесс фосфоролиза в целом, тонко регулируется.
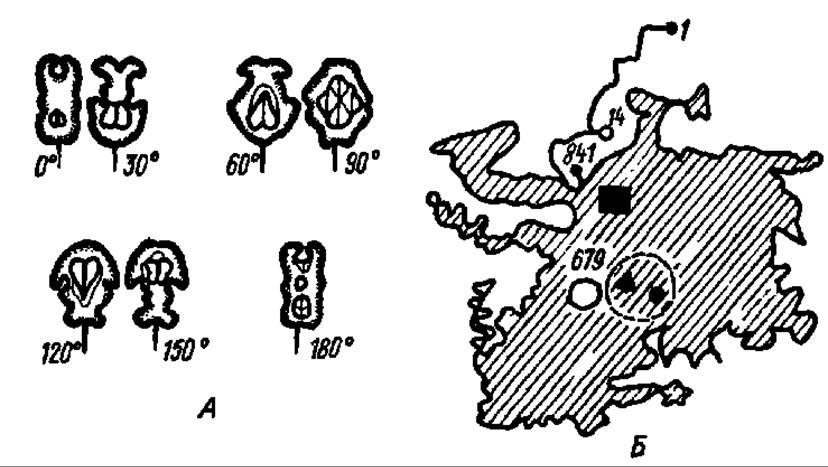
Рис. 107. Четвертичная структура гликогенфосфорилазы мышц кролика (А) и третичная структура протомера (В):
А — при вращении модели от 0-до 160° ясно прослеживается наличие четырех протомеров и их взаимное расположение в мультимере; Б — детали пространственного расположения полипептидной цели опущены и показан лишь общий контур протомера, 1 — N-концевая аминокислота; 841 — С-концевая аминокислота; 14 — остаток серинфосфата; 679 — остаток лизина с присоединенным к нему пиридоксальфосфатом; пунктирным кружком обозначена локализация активного центра, косой штриховкой — субстратного центра, черным прямоугольником — АМФ-связывающий центр (АМФ — активатор фермента), треугольником и точкой в зоне активного центра — места связывания нуклеозидов и глюкозы (оба — ингибиторы)
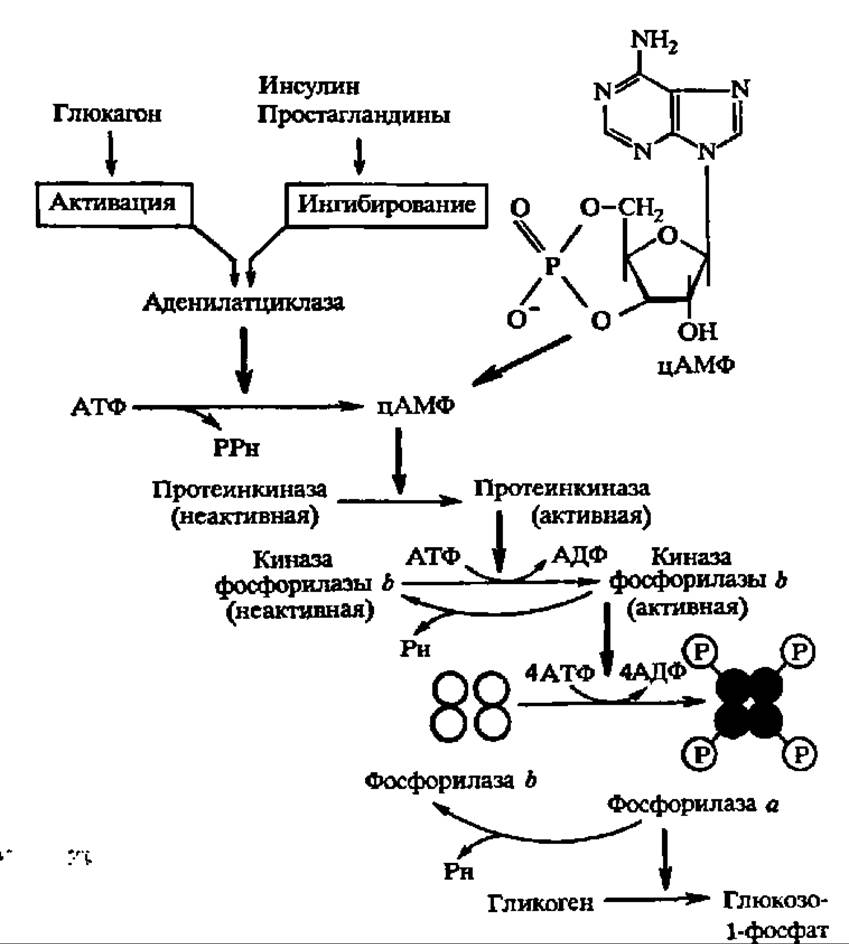
Рис. 108. Регуляция активности гликогенфосфорилазы при посредстве протеинкиназных и протеинфосфатазных реакций (пояснение в тексте)
В последние годы появились новые данные о структуре киназы фосфорилазы b и регуляции ее активности. Полагают, что ее субчастица (М = 316 000) состоит из 4 протомеров (а — 138000; ß — 117000; у — 44000 и 5 — 17000), a полная молекула — из 4 субчастиц (М = 1 250000). Каталитическую функцию несет протомер у, регуляторную — протомеры а, ß и δ, причем последний является Ca2+-зависимым и отождествляется в Са2+-связывающим регуляторным белком кальмодулином.
Превращение моносахаридов. Как показано выше, при распаде олиго- и полисахаридов возникают свободные монозы и их фосфорные эфиры. Дальнейший обмен моносахаридов идет такими путями, что используются только фосфорные эфиры моносахаридов, свободные же монозы фосфорилируются, т. е. превращаются в фосфорные эфиры.
Фосфорилирование свободных моносахаридов — обязательная реакция на пути их использования для нужд организма. Оно приводит к возникновению более реакционноспособных, чем свободные моносахариды, фосфорных эфиров моносахаридов и поэтому часто рассматривается как реакция активирования.
Фосфорилирование моносахаридов осуществляется при взаимодействии их с АТФ и ускоряется специфическими (и неспецифическими) фосфотрансферазами, которые называются также киназами. Фосфорилирование глюкозы, например, идет согласно схеме:
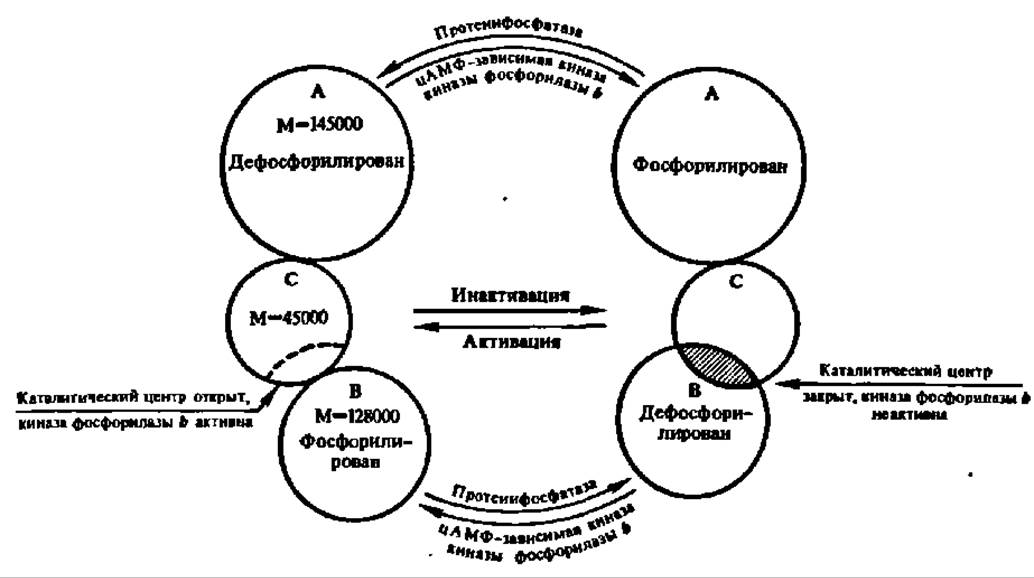
Рис. 109. Регуляция активности киназы фосфорилазы b (пояснение в тексте)
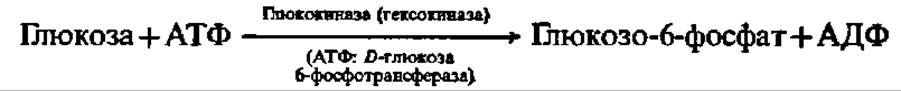
Уравнение этой реакции приведено выше (см. с. 124).
В природе обнаружено более двух десятков индивидуальных фосфотрансфераз, переносящих остатки фосфорной кислоты с АТФ на те или иные моносахариды. Молекулы многих из них, как выяснено в последнее время, построены из 2 субъединиц. Так, гексокиназа из дрожжей при М = 102 000 состоит из 2 субъединиц с М = 51000 каждая и обнаружено 2 ее изозима; галактокиназа из эритроцитов человека при М = 53000 — из 2 субъединиц по 27000 каждая; рибулокиназа из кишечной палочки при М = 98 000 — из 2 субъединиц по 50000 каждая и т. п. Изучение третичной структуры субъединицы дрожжевой гексокиназы выявило её двухдоменную структуру и локализацию связывания АТФ и глюкозы на дне щели между доменами, где, собственно, и протекает процесс переноса фосфата при непременном участии Mg2+. Выяснены и более тонкие детали этого процесса, в частности переход субъединицы из открытой конформации в момент сорбции субстратов в закрытую в процессе осуществления реакции (рис. 110).
Рис. 110. Конформационные изменения субъединицы молекулы гексокиназы в процессе возникновения фермент-субстратного комплекса:
А — до присоединения глюкозы (открытая конформация); Б — после присоединения глюкозы (закрытая конформация). Достаточно легко различимы два главных домена субъединицы, перемещение которых обеспечивает изменение ее конформации
Рассмотрим еще несколько примеров действия киназ. Так, фруктокиназа ускоряет реакцию образования фруктозо-6-фосфата:
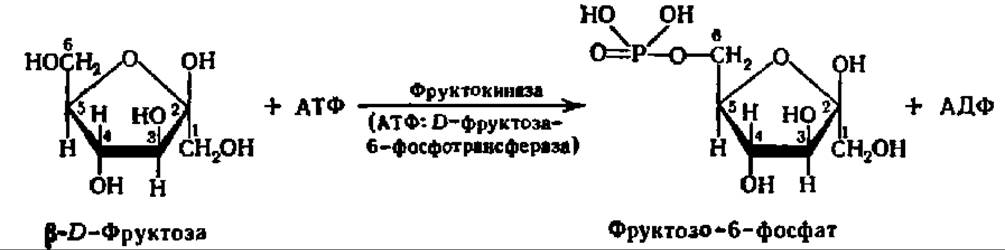
Аналогично протекает взаимодействие между рибозой и АТФ:
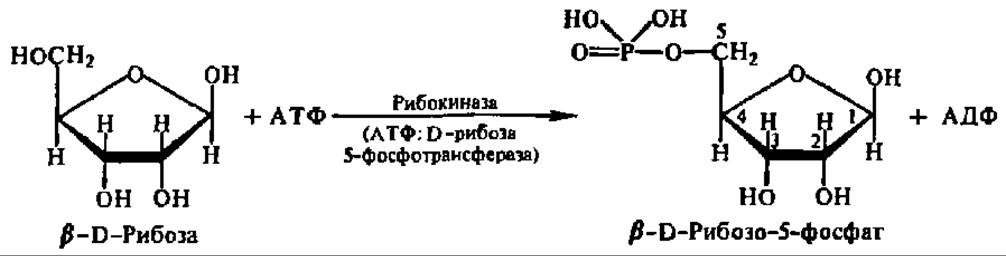
Следовательно, практически любой моносахарид может быть переведен в фосфорный эфир. Именно в виде фосфорных эфиров и осуществляется дальнейший обмен моносахаридов.
Важнейшей особенностью фосфорных эфиров моносахаридов является их Способность к изомеризация. Последняя может осуществляться как за счет стереоизомеризации (т. е. изменения пространственного расположения атомов и атомных групп в молекуле), так и в результате внутримолекулярного переноса атомов Н. Одним из примеров подобного рода превращений служит переход глюкозо-6-фосфата в фруктозо-6-фосфат, интенсивно осуществляемый в мышечной ткани и приводящий к установлению равновесия между этими двумя эфирами в отношении 2:1:
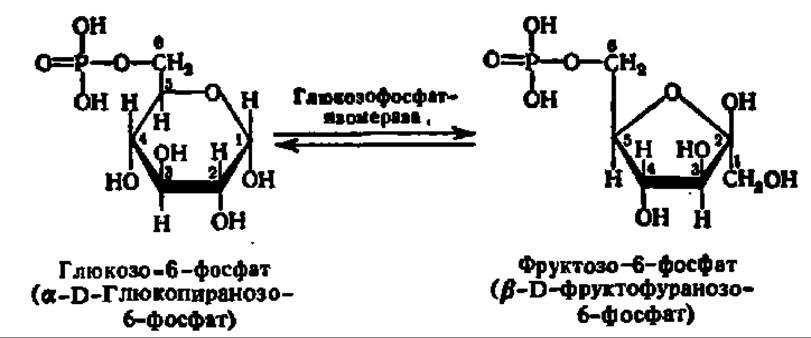
Приведем еще некоторые характерные в этом смысле реакции. Так, рибозо-5-фосфат может превращаться в рибулозо-5-фосфат, а последний, в свою очередь, — в ксилулозо-5-фосфат:
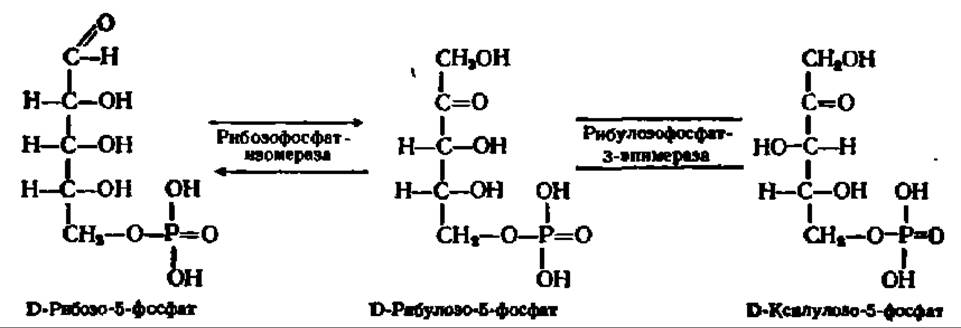
В первом случае происходит внутримолекулярный перенос водорода, во втором — изменение расположения в пространстве атома Н и ОН-группы при 3-м углеродном атоме. И тот и другой процессы ускоряются ферментами, принадлежащими к классу изомераз.
Особенно легко процесс перестройки одного моносахарида в другой протекает, если фосфорный эфир моносахарида соединен с уридин-5-фосфатом. Примером может служить превращение галактозы в глюкозу и обратно:
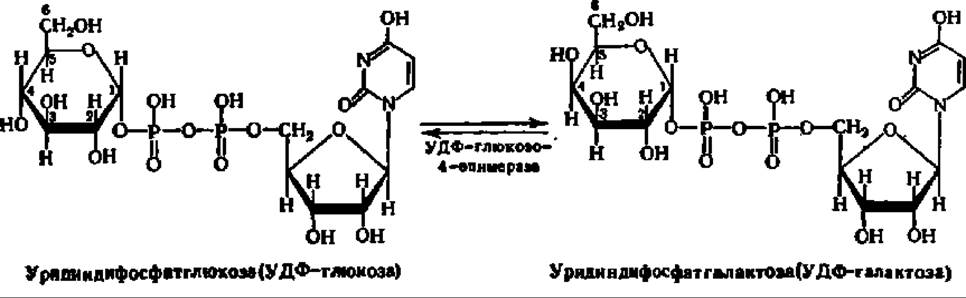
Если учесть, что в организме не только фосфорные эфиры моносахаридов, но и свободные моносахариды могут превращаться друг в друга, то появляется возможность перехода от любой гексозы или пентозы к любой другой, изомерной ей. Это имеет огромное значение. В природе и синтез и распад моносахаридов, как и весь процесс обмена углеводов, протекает через некоторые их формы, занимающие в данном процессе ключевые позиции. Это, в первую очередь, фосфорные эфиры двух моносахаридов: глюкозо-6-фосфат и рибулозо-5-фосфат.
Обмен глюкозо-6-фосфата. Глюкозо-6-фосфат образуется в организме разными путями. Во-первых, он может синтезироваться путем фосфорилирования глюкозы за счет ее взаимодействия с АТФ. Во-вторых, он образуется в результате реакции изомеризации фосфорных эфиров других изомерных ему гексозофосфорных эфиров. В-третьих, он получается из глюкозо-1-фосфата, который представляет собой продукт фосфоролиза олиго- и полисахаридов. Две первые реакции рассмотрены в предыдущем разделе. Что касается преобразования глюкозо-1-фосфата в глюкозо-6-фосфат, то эта реакция протекает в два этапа при участии фермента фосфоглюкомутазы. Молекулярная масса фермента из мышц кролика равна 62000; молекула фосфоглюкомутазы состоит из двух субъединиц с М = 31000 каждая. Активный центр ее включает в свой состав остаток фосфосерина, с которого остаток фосфорной кислоты передается на глюкозо-1-фосфат с образованием глюкозо-1,6-дифосфата и дефосфорилированного фермента. Последний, взаимодействуя с глюкозо-1,6-дифосфатом, снова превращается в фосфопротеин, однако получает остаток фосфорной кислоты, присоединённой к 1-му углеродному атому глюкозы с высвобождением соответственно глюкозо-6-фосфата:
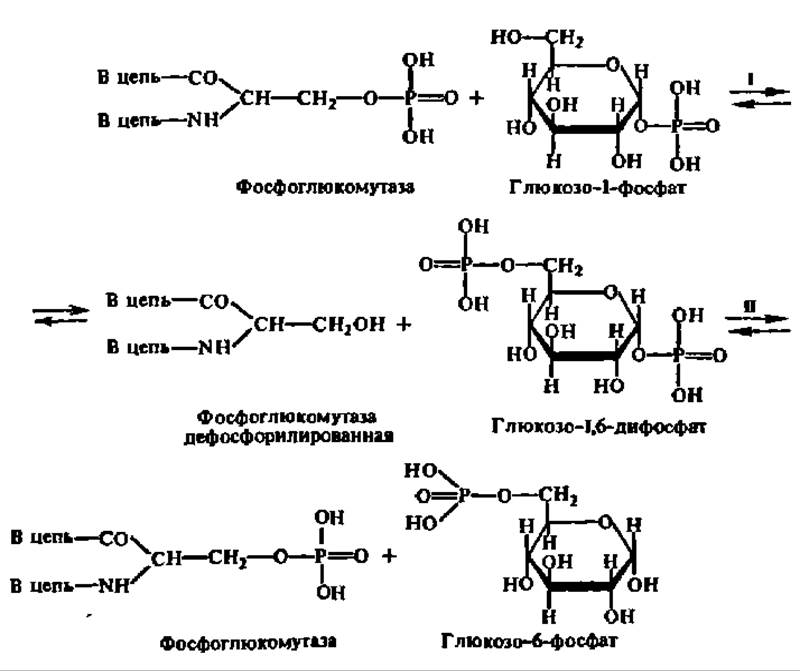
Равновесие рассмотренной реакции сильно сдвинуто в сторону образования глюкозо-6-фосфата. Поэтому в тканях содержание глюкозо-1-фосфата не превышает 3—4% от общего количества гексозомонофосфатов в организме.
Ілюкозо-6-фосфат подвергается в организме разнообразным превращениям. Некоторая доля его распадается в конечном счете до СО2 и Н2О. При этом многократно повторяются реакции окисления (дегидрогенизации) как самого глюкозо-6-фосфата, так и продуктов его дальнейшего распада. Сопряженно с передачей атомов водорода, снятых с глюкозо-6-фосфата и возникших из него субстратов, на кислород (с образованием молекул воды) осуществляется синтез АТФ из АДФ и неорганического фосфата, т. е. запасается, аккумулируется энергия в составе макроэргических связей молекул АТФ. Кроме того, некоторое количество молекул АТФ синтезируется здесь же иным путем. Следовательно, распад глюкозо-6-фосфата служит энергетическим целям: является источником энергии для организма.
Вместе с тем значительная часть промежуточных продуктов, возникающих в процессе обмена глюкозо-6-фосфата, используется для синтеза аминокислот (белков), нуклеотидов (нуклеиновых кислот), глицерина и высших жирных кислот (триглицеридов, фосфатидов), стеролов (стеридов) и т. п. В частности, как описано выше, для синтеза аланина используется пировиноградная кислота (см. с. 276), являющаяся непременным промежуточным продуктом при распаде глюкозо-6-фосфата по дихотомическому пути (см. следующий раздел). Другие промежуточные продукты распада глюкозо-6-фосфата: 3-фосфоглицериновая кислота и фосфоенолпировиноградная кислота (см. ниже) идут на синтез фенилаланина, тирозина, триптофана и серина: Включаясь в цикл трикарбоновых и дикарбоновых кислот (см. рис. 117), пировиноградная кислота, превращаясь в щавелевоуксусную и а-кетоглутаровую, дает начало аспарагиновой и глутаминовой кислотам, а из них — ряду других аминокислот. Рибозо-5-фосфат, образующийся при апотомическом распаде глюкозо-6-фосфата (см. схему 6), служит для синтеза гистидина и, в еще большей степени, для синтеза пиримидиновых и пуриновых нуклеотидов (см. гл. VI).
Таким образом, глюкозо-6-фосфат обеспечивает организм и энергией и строительным материалом для синтеза новых органических соединений, используемых в процессе жизнедеятельности.
Распад глюкозо-6-фосфата осуществляется преимущественно двумя путями. В одном случае на определенной стадии происходит распад шестиуглеродной молекулы на две трехуглеродные, т. е. пополам. Этот путь получил название дихотомического распада. Второй путь состоит в потере глюкозо-6-фосфатом 1-го углеродного (головного) атома и именуется апотомическим распадом. Есть еще третий путь, содержащий элементы первого и второго. Рассмотрим каждый из них.
Дихотомический путь распада глюкозо-6-фосфата. Вступая на дихотомический путь распада, глюкозо-6-фосфат прежде всего претерпевает изомеризацию и превращается в фруктозо-6-фосфат, который далее фосфорилируется по 1-му углеродному атому, образуя фруктозо-1,6-дифосфат:
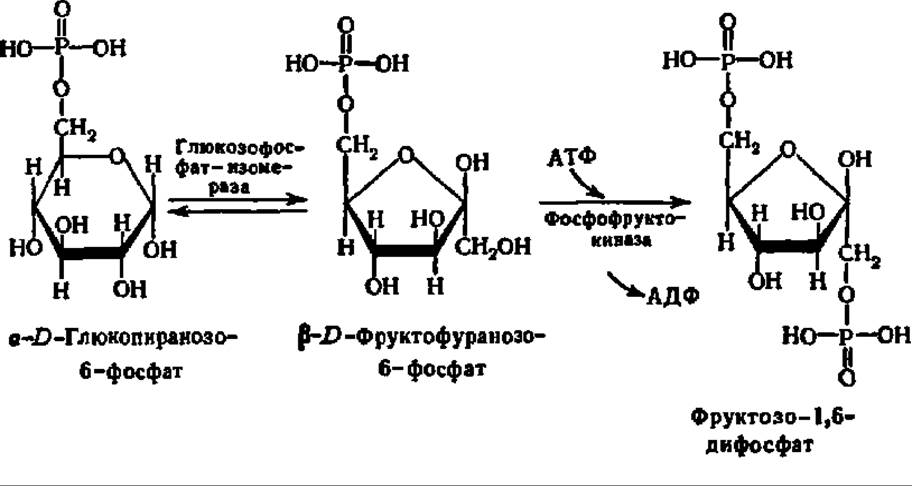
Фосфофруктокиназа, представляющая собой белок с М = 140000 у бактерий и от 360000—400000 (во всех случаях — тетрамер) до 800000 (гексамер или октамер) у эукариот, является самым «медленным» из всех ферментов, обслуживающих дихотомический распад углеводов. Эта практически необратимая реакция лимитирует весь процесс. В то же время активность глюкозофосфатизомеразы, например, в дрожжах, в 500 раз превышает активность фосфофруктокиназы и на 1—2 порядка выше, чем у других ферментов дихотомического распада, т. е. это один из самых «быстрых» ферментов обмена глюкозо-6-фосфата. Строение и механизм действия фосфофруктокиназы из термофильной бактерии детально изучены (рис. 111).
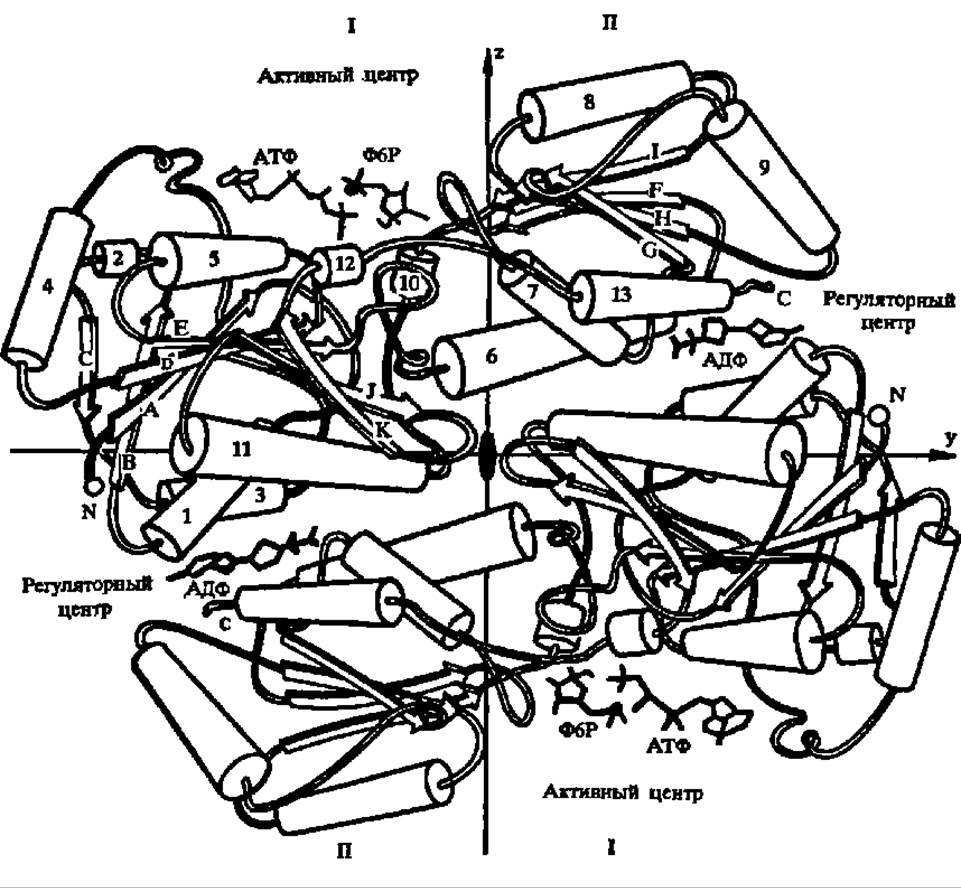
Рис. 111. Строение половины молекулы фосфофруктокиназы из термофильной бактерии (3-я и 4-я субъединицы не показаны)
Молекулярная масса субъединицы, составленной из 316 аминокислотных остатков, равна 33 900; третичная структура характеризуется наличием ß-слоев (обозначены А—К) и а-спиралей (обозначены 1—13), а также существованием двух доменов (I и II); на границе доменов располагаете! активный центр, связывающий фруктозо-6-фосфат, АТФ и Mg2+; вблизи С-концевой аминокислоты II домена находится аллостерический центр (АДФ — активатор, фосфоенолпировиноградная кислота — ингибитор), обеспечивающий регуляцию активности фосфофруктокиназы при посредстве ряда эффекторов, что является важнейшей особенностью этого фермента. В эпимолекуле между субъединицами есть полость диаметром 0,7 им, т. е. тетрамерный фермент имеет трубчатую структуру. Ф6Р — фруктозо-6-фосфат
Концентрация фруктозо-1,6-дифосфата поддерживается на строго определенном уровне при посредстве сложного комплекса регуляторных процессов: снижается под действием фруктозо-1,6-дифосфатазы (М = 140000, 4 X 35 000) и возрастает под влиянием фосфофруктокинаэы. Однако активность первой тормозится, а второй — побуждается в присутствии фруктозо-2,6-дифосфата, который, в свою очередь, синтезируется в печени при посредстве 6-фосфофрукто-2-фосфокиназы. Более того, последняя реакция цАМФ-зависима, а сам этот фермент в печени бифункционален: один его домен ускоряет синтез, а другой — распад фруктозо-2,6-дифосфата (рис. 112). В дрожжах 6-фосфофрукто-2-фосфокиназа и фруктозо-2,6-дифосфатаза не образуют бифункционального фермента и являются самостоятельными энзимами.
Последнее соединение (фруктозо-1,6-дифосфат) и подвергается далее дихотомическому распаду на две фосфотриозы, превращающиеся друг в друга:
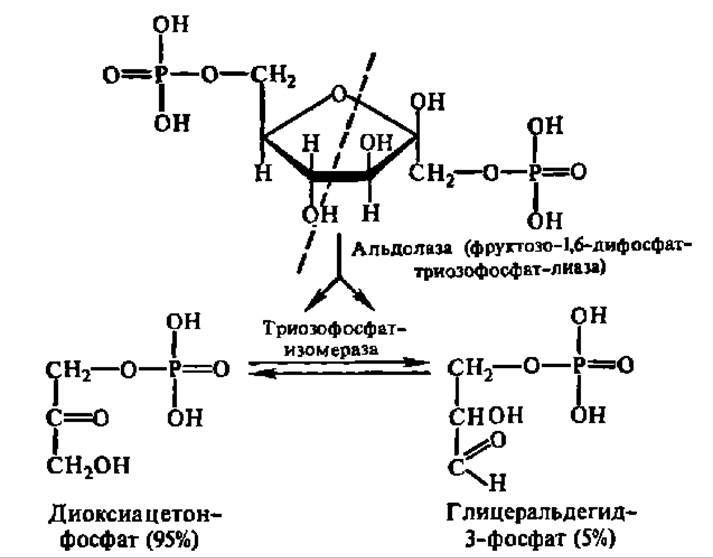
Оба фермента, ускоряющие приведенные выше реакции, получены в кристаллическом состоянии. Альдолаза из мышц кролика характеризуется М = 160000 (4x40000), а триозофосфат-изомераза — 53000 (2x26500). Выяснена первичная структура последней и параметры ее вторичной структуры (52% а-спиралей и 24% ß-слоев). Хотя при дихотомическом расщеплении фруктозо-1,6-дифосфата (видимо, расщепляется его открытая форма) получается равное количество обеих фосфотриоз, в состоянии равновесия между ними преобладает фосфодиоксиацетон.
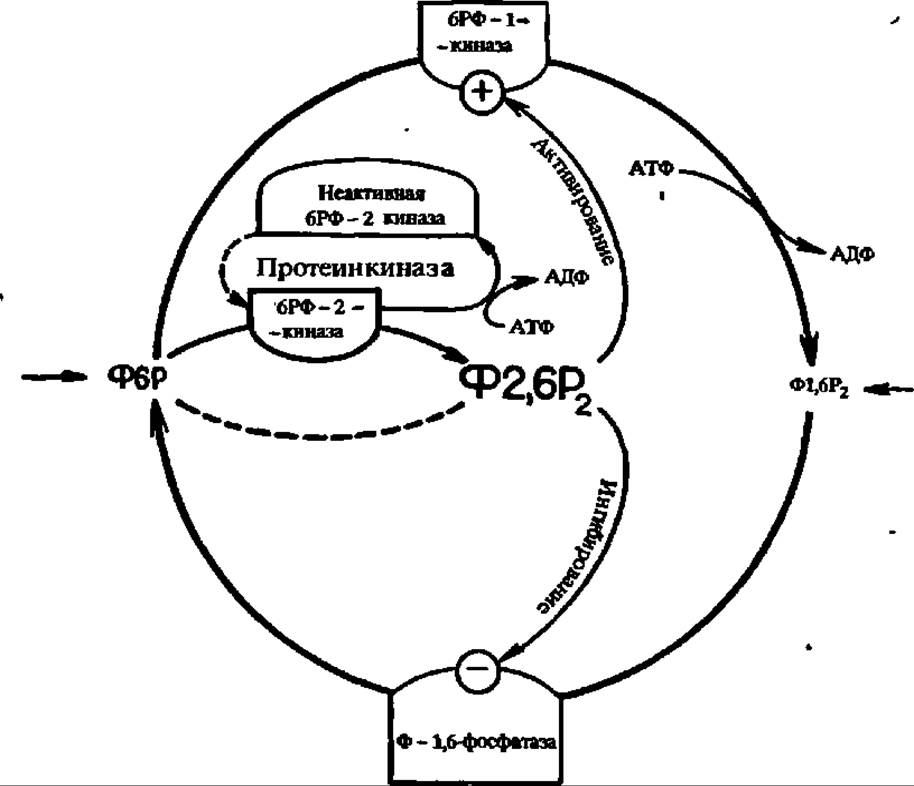
Рис. 112. Регуляция содержания ключевого метаболита дихотомического распада углеводов — фруктозо-1,6-дифосфата (Ф1,6Р2):
6РФ-1-киназа — фруктозо-6-фосфат-1-киназа; 6РФ-2-киназа — фруктозо-6-фосфат-2-киназа; Ф6Р — фруктозо-6-фосфат; Ф2,6Р2 — фруктозо-2,6-дифосфат; Ф1,6Р2-фосфатаза — фруктозо-1,6-дифосфатаза (Остальные пояснения в тексте)
В дальнейший обмен вступает только 3-фосфоглицериновый альдегид. По мере расходования убыль этого соединения восполняется за счет фосфодиоксиацетона, который практически нацело в него превращается. Следовательно, из каждой молекулы фруктозо-1,6-дифосфата фактически возникает две молекулы 3-фосфоглицеринового альдегида, претерпевающего далее распад в соответствии со следующей схемой:
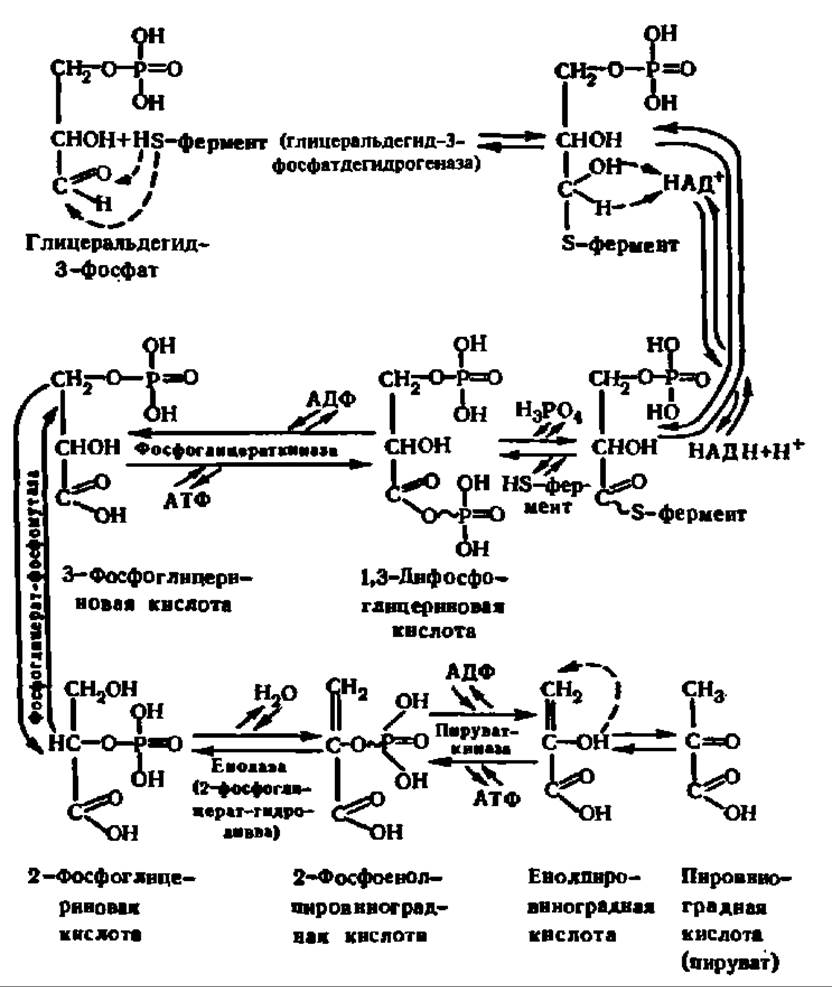
Схема 5. Обмен 3-фосфоглицеринового альдегида
Все реакции, происходящие при обмене 3-фосфоглицеринового альдегида, осуществляются ферментативным путем. Характерная их особенность состоит в том, что на каждую молекулу распадающегося 3-фосфоглицеринового альдегида синтезируются две молекулы АТФ из АДФ и остатков фосфорной кислоты, поступающих сначала от 1,3-дифосфоглицериновой кислоты, а затем от 2-фосфоенолпировиноградной кислоты (схема 5). Таким образом, уже здесь запасается энергия, выделяющаяся в процессе постепенного окисления фосфоглицеринового альдегида. Конечным продуктом распада глюкозо-6-фосфата является пировиноградная кислота (ПВК). В зависимости от объекта и условий, в которых идет обмен углеводов, дальнейшая ее судьба различна (см. ниже).
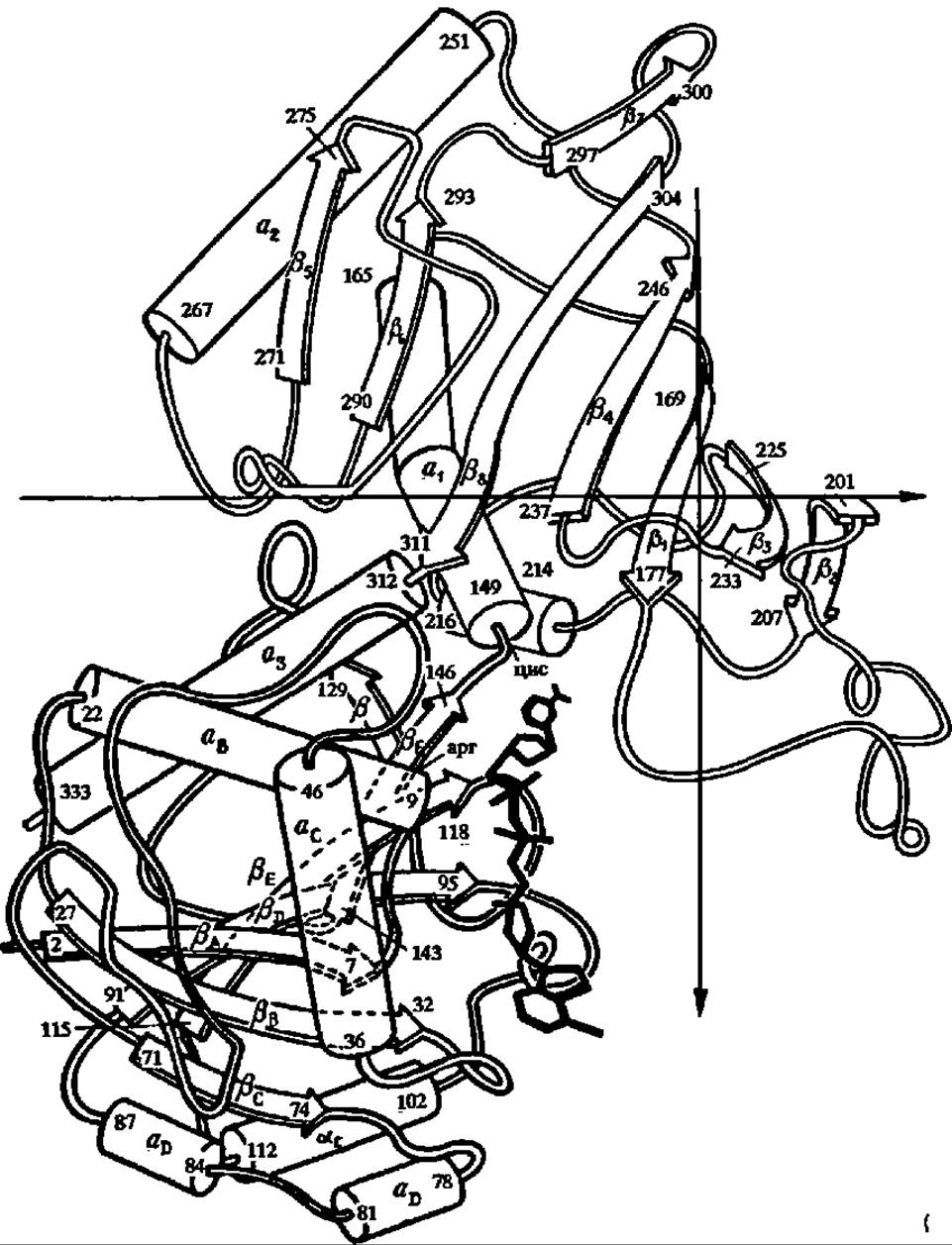
Рис. 113. Структура субъединицы глицеральдегид-3-фосфатдегидрогеназы:
а-спирали обозначены цилиндрами; фрагменты полипептидной цепи в ß-слоях — стрелками; в верхней части рисунка нумерация их числовая (а1, а2 и т. д.; β1, ß2 и т. д), a в нижней, в области нуклеотидсвязывающего домена и активного центра — буквенная (аA, аВ и т. д.; ßA, ßВ и т. д.); здесь же толстыми линиями показан кофермент — никотинамидадениндинуклеотид (НАД), причем его восстанавливаемая никотинамидная Часть сближена с цис149, к которому присоединяется субстрат, что создает необходимые условия для окисления последнего
Наиболее сложной из всех приведенных выше реакций на пути от 3-фосфоглицеринового альдегида до ПВК является реакция окисления фосфоглицеринового альдегида в фосфоглицериновую кислоту. Остановимся на ней несколько подробнее. Реакция ускоряется глицеральдегид-3-фосфатдегцдрогеназой, полученной в кристаллическом состоянии из дрожжей, термофильных бактерий и мышц кролика, омара и свиньи. Молекулярная масса фермента в большинстве случаев равна 144000. Молекула фермента состоит из четырех субъединиц с М = 37 000. Первичная структура их выяснена у глицеральдегид-3-фосфатдегидрогеназы из пекарских дрожжей, термофильной бактерии и мышц свиньи и омара, а третичная — у фермента из термофильной бактерии и мышц омара (рис. 113).
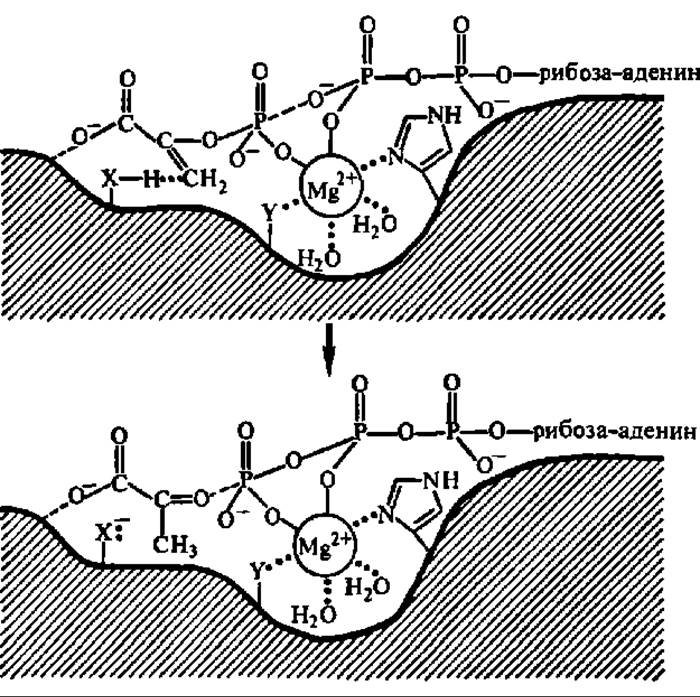
Рис. 114. Механизм пируваткиназной реакции при сопряжении окисления с фосфорилированием на уровне субстрата
Ион металла участвует в образовании тройного мостикового комплекса (фосфоенолпируват — металл—АДФ), обеспечивающего фосфорилирование АДФ; в активном центре фермента координирующей функции иона металла способствует радикал гис, а превращению енолпировиноградной кислоты в пировиноградную — донор протонов ХН; ион металла закрепляется в активном центре также радикалом У и гидратируется. Препараты пируваткиназы, выделенные из разных источников, существенно не отличаются по молекулярной массе (она близка к 200000); молекула фермента в подавляющем большинстве случаев тетрамерна. Третичная структура субъединицы мышечной пируваткиназы характеризуется наличием трех доменов, на границе двух из них расположен активный центр
Каждая субъединица несет одну молекулу НАД+ и 4 свободные HS-группы, принадлежащие остаткам цистеина. Сначала фосфоглицериновый альдегид присоединяется к ферменту по радикалу остатка цистеина, занимающего 149-е положение в полипептидной цепи; в активный центр фермента входят также остаток аргинина, находящийся в 231-м положении, и остаток гистидина, занимающий 176-ю позицию. Затем в действие вступает НАД+, отнимающий от субстрата атом водорода в виде гидрид-иона (схема 5), и гис176 активного центра, снимающий другой атом водорода с ОН-группы тиополуацеталя в виде протона; радикал гистидина удерживает снятый протон в течение непродолжительного времени и потом высвобождает его в среду (схема 5 и рис. 113).
В этот момент связь между остатком 3-фосфоглицериновой кислоты и ферментом становится макроэргической (обозначена значком ~, схема 5). Указанная связь спонтанно распадается в присутствии Н3РО4с образованием 1,3-дифосфоглицериновой кислоты. Остальные реакции идут в основном при участии соответствующих киназ. Важно отметить, что в момент отщепления воды от 2-фосфоглицериновой кислоты (схема 5) также возникает макроэргическая связь у остатка фосфата, что делает возможной дальнейшую киназную реакцию с образованием АТФ. Такой путь биосинтеза АТФ называется субстратным фосфорилированием, которое возможно лишь потому, что в активном центре фосфоглицераткиназы и пируваткиназы при участии Mg2+ сближаются концевой фосфат АДФ и переносимый на него фосфат, связанный макроэргической связью в 1,3-дифосфоглицериновой или 2-фосфоенолпировиноградной кислоте. В частности, эта важнейшая реакция фосфорилирования АДФ на уровне окисляемого субстрата детально изучена у пируваткиназы (рис. 114).
Апотомический путь распада глюкозо-6-фосфата. При апотомическом распаде глюкозо-6-фосфата не происходит его превращения в фруктозо-1,6-дифосфат в результате введения в молекулу второй фосфатной группы. Распад глюкозо-6-фосфата в этом случае начинается реакцией окисления его в 6-фосфоглюконолактон. Окисление состоит в отнятии двух атомов водорода от 1-го углеродного атома глюкозо-6-фосфата. Акцептором Н служит НАДФ+, являющийся коферментом глюкозо-6-фосфатдегидрогеназы, ускоряющей эту реакцию:
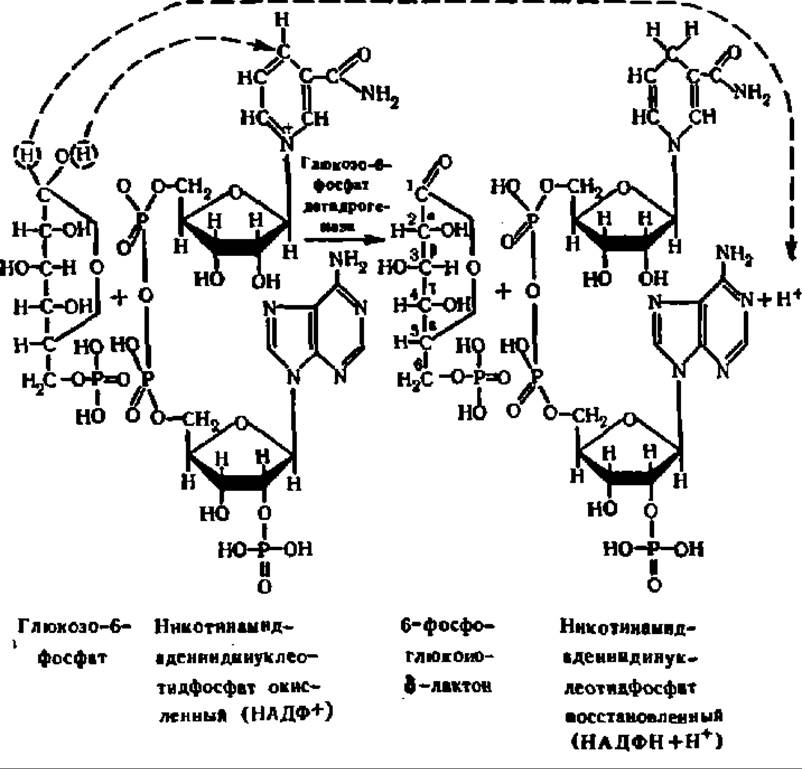
Глюкозо-6-фосфатдегидрогеназа, открытая более полувека тому назад О. Варбургом и сотр. и В. А. Энгельгардтом и А. П. Бархаш, выделена из различных источников и характеризуется либо димерной, как, например, в молочной железе крысы: М = 130000 (2x63000) или эритроцитах человека: М = 204000 (2x101400), либо тетрамерной, как, например, у нейроспоры: М = 206 000 (4 x 57000), в надпочечниках быка: М = 284 000 (4 x 64000) или грене тутового шелкопряда: М = 232 000 (4x54000), структурой. Она существует в виде множественных форм, которыми особенно богаты эритроциты человека, а ее активность задается соотношением НАДФ+/НАДФН. Выяснена первичная структура глюкозо-6-фосфатдегидрогеназы из Bacillus megaterium (М = 118 000; 4 X 29 500); в ее субъединице — 262 аминокислотных остатка.
б-Фосфоглюконолактон при участии фермента глюконолактоназы гидролизуется до 6-фосфоглюконовой кислоты, которая претерпевает окислительное декарбоксилирование и превращается в рибулозо-5-фосфат:
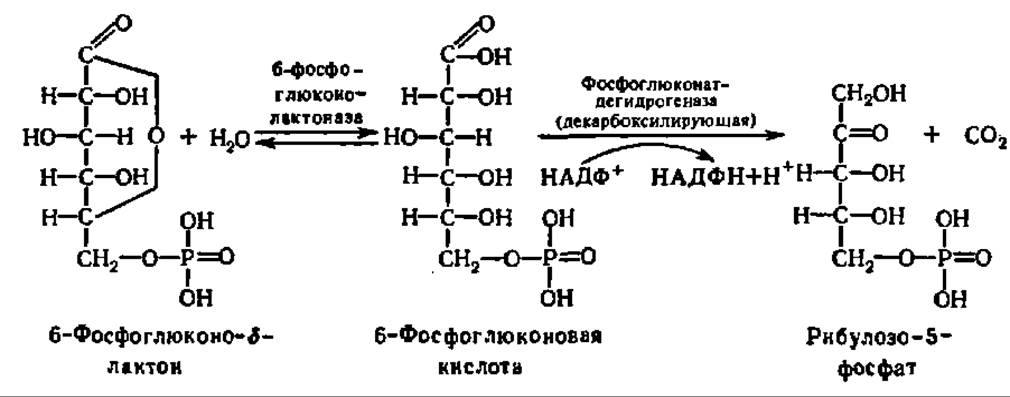
Фосфоглюконатдегидрогеназа (декарбоксилирующая) представлена белком с М = 100000, состоящим из двух равных субъединиц, независимо от источника выделения (печень овцы и крысы, эритроциты человека, дрожжи).
Дальнейший обмен рибулозо-5-фосфата — одного из центральных веществ в углеводном обмене — протекает весьма сложно (схема 6). Многократно изомеризуясь, в частности, переходя в рибозо-5-фосфат и ксилулозо-5-фосфат, а также вступая в транскетолазные и трансальдолазные реакции, заключающиеся в переносе двууглеродных и трехуглеродных фрагментов от одного фосфорного эфира к другому, рибулозо-5-фосфат снова превращается в глюкозо-6-фосфат. Подсчитано, что из 6 молекул рибулозо-5-фосфата получается 5 молекул глюкозо-6-фосфата. Таким образом, суммарный эффект всех реакций, осуществляющихся при апотомическом распаде глюкозо-6-фосфата, сводится к тому, что из каждых 6 его молекул одна полностью распадается. Это можно выразить следующим уравнением:
6 Молекул глюкозо-6-фосфата+12 НАДФ+ + 7Н2О → 6СО2 + 12 НАДФН+
+12Н+ + Н3РО4 + 5 молекул глюкозо-6-фосфата
После сокращения 5 молекул глюкозо-6-фосфата в левой и правой частях уравнения остается следующее:
Глюкозо-6-фосфат+12 НАДФ+ +7Н2О → 6СО2 +12 НАДФН +
+ Н3РО4+12Н+
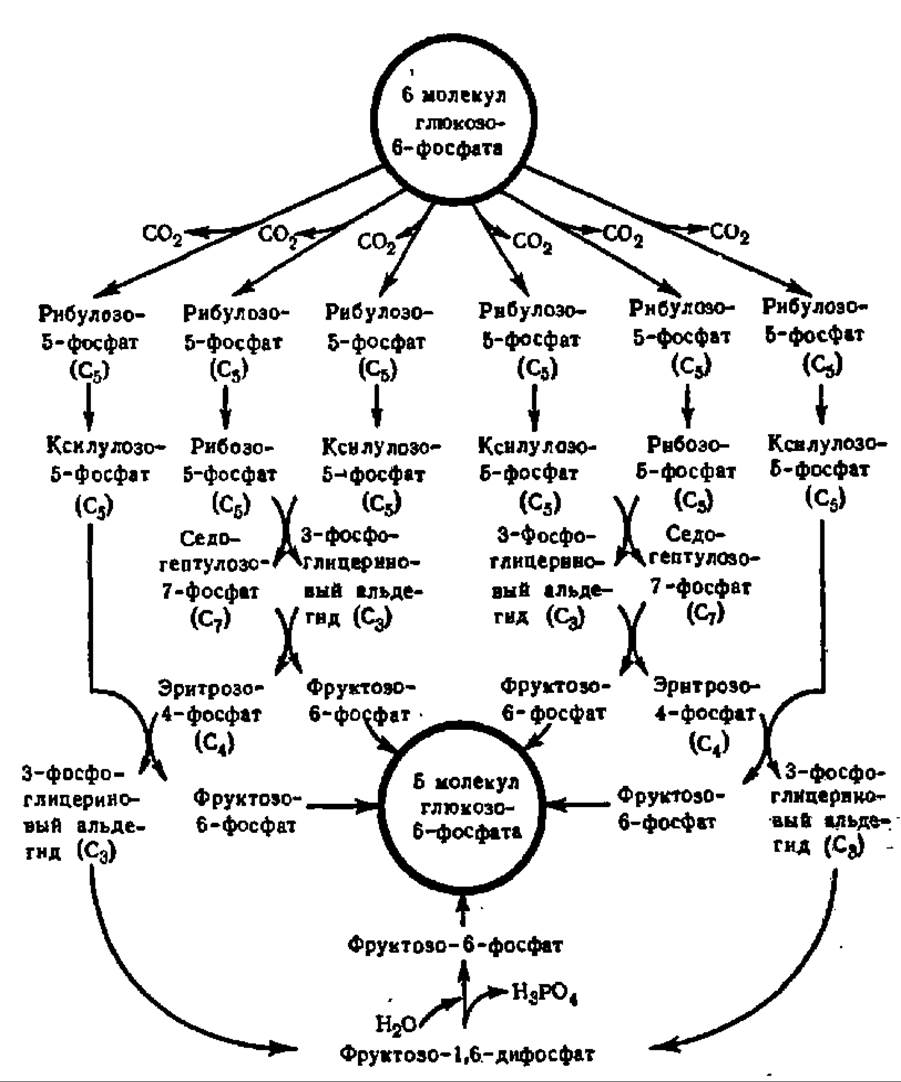
Схема 6. Превращения глюкозо-6-фосфата при апотомическом распаде (пояснения в тексте) исходя из распределения 14С-промежуточных соединений в экстрактах ацетоновых порошков печени крысы и листьев и корешков гороха (В. Хоррекер и др., 1951). В 1978—1983 гг. усложнена Дж. Вильямсом и др. введением в нее арабинозо-5-фосфата, диоксиацетонфосфата и октулозо-1,8-дифосфата в качестве метаболитов, которые на схеме не показаны
Как следует из схемы 6, важнейшее значение в апотомическом распаде углеводов имеет превращение фосфопентоз. Поэтому этот путь обмена углеводов называют также пентозофосфатным циклом. Центральной реакцией в нем является перенос двууглеродных фрагментов, осуществляемый при каталитическом воздействии транскетолазы. Этот фермент (у нас в стране) детально изучен Г. А. Кочетовым с сотр. Транскетолаза из пекарских дрожжей (М = 160 000) построена из двух субъединиц (2x75000), каждая из которых содержит в качестве кофермента тиаминпирофосфат, присоединенный к белковой части соответственно через радикал триптофана и пирофосфатную группировку (рис. 115). Именно при посредстве тиаминпирофосфата и осуществляется перенос двууглеродных фрагментов. Он идет с помощью  -группы тиазолового цикла (рис. 115). В каждой из субъединиц 20% а-спиралеи, 40% β-слоев и 40% полипептидной цепи в виде клубка. Для становления димерной формы фермента и его функции важен Са2+. Транскетолаза из эритроцитов человека резко отличается от дрожжевой. Будучи очищена в 70000 раз, она имеет М = 140 000 и не нуждается ни в тиаминпирофосфате, ни в Mg2+ для проявления активности.
-группы тиазолового цикла (рис. 115). В каждой из субъединиц 20% а-спиралеи, 40% β-слоев и 40% полипептидной цепи в виде клубка. Для становления димерной формы фермента и его функции важен Са2+. Транскетолаза из эритроцитов человека резко отличается от дрожжевой. Будучи очищена в 70000 раз, она имеет М = 140 000 и не нуждается ни в тиаминпирофосфате, ни в Mg2+ для проявления активности.
Постепенно укрепляется мнение, что транскетолазные и другие трансферазные реакции в обмене углеводов являются одними из наиболее древних. По мере эволюции к ним присоединились дегидрогеназные процессы (см. выше две первые реакции окисления в апотомическом распаде углеводов) и апотомический путь обмена углеводов приобрел законченный вид. Но в дальнейшем дихотомический путь распада углеводов стал преобладающим, и сейчас пентозофосфатный цикл в целом, особенно у высших животных, занимает скромное место.
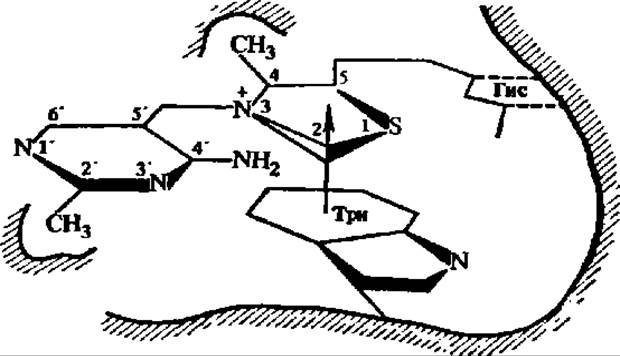
Рис. 115. Схема связывания тиаминпирофосфата с апотранскетолазой в активном центре фермента (пояснения в тексте)
Путь Этнера—Дудорова. Кроме дихотомического и апотомического путей обмена глюкозо-6-фосфата существует еще один путь, характерный для микроорганизмов (у некоторых из них при его посредстве распадается от 30 до 50% глюкозы), названный в честь его первооткрывателей (1952). Первые стадии распада глюкозо-6-фосфата по этому пути, вплоть до образования 6-фосфоглюконовой кислоты, полностью повторяют апотомический путь. Но далее 6-фосфоглюконовая кислота окисляется без декарбоксилирования в 2-кето-6-фосфоглюконовую кислоту, которая, восстанавливаясь по 3-му углеродному атому, переходит в 2-кето-3-дезокси-6-фосфоглюконовую кислоту, претерпевающую альдолазное расщепление:
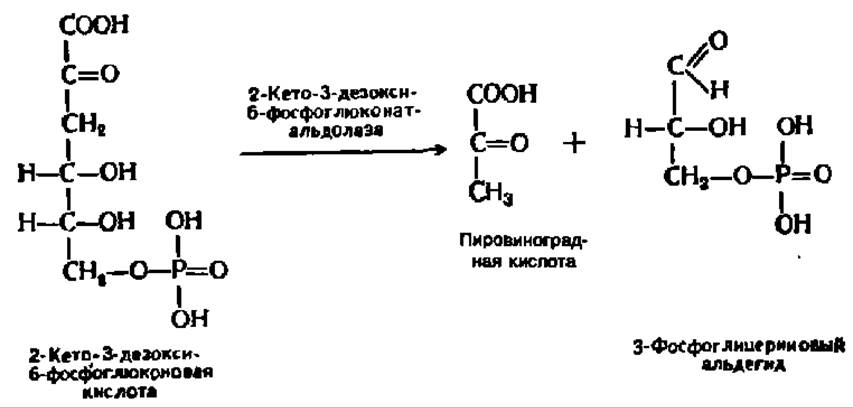
Оба конечных продукта распада поступают в общий метаболический фонд и подвергаются обычным превращениям.
Обмен пировиноградной кислоты (ПВК). Проследим теперь судьбу ПВК, возникающей в качестве конечного продукта при дихотомическом распаде глюкозо-6-фосфата и другими путями. В зависимости от места и условий протекания процесса в организме, наличия или отсутствия в последнем тех или иных ферментных систем и т. п. судьба эта различна.
Если процесс идет в анаэробных условиях или при недостаточном снабжении кислородом, то простейший вариант обмена ПВК заключается в ее восстановлении до молочной кислоты. Донором атомов водорода при этом служит НАДН, образующийся в процессе окисления 3-фосфоглицеринового альдегида при дихотомическом распаде глюкозо-6-фосфата (см. схему 5) и во многих других случаях. Эта реакция ускоряется лактатдегидрогеназой. На этом ферменте впервые детально был разработан вопрос об изозимах (см. с; 101). Изозим типа MMMM характерен для анаэробных тканей и обеспечивает процесс превращения ПВК в молочную кислоту; изозим типа НННН локализован в тканях с высоким аэробиозом и превращает в них молочную кислоту в ПВК. Гибридніе формы (НМММ, ННММ и НННМ) обладают промежуточной активностью. Так достигается очень тонкая регулировка направления ферментативного процесса и соотношения в тканях молочной и пировиноградной кислот.
Таким образом, в анаэробных условиях каждая молекула глюкозо-6-фосфата дает две молекулы молочной кислоты, которая в этом случае представляет конечный продукт реакции. Если исходным углеводом для образования глюкозо-6-фосфата, а затем молочной кислоты служит глюкоза, то процесс называют гликолизом. Если же исходным углеводом, дающим начало глюкозо-6- фосфату (через глюкозо-1-фосфат) и потом молочной кислоте, является гликоген, то процесс называют гликогенолизом. Учитывая, что и в том и в другом случае на промежуточных стадиях дихотомического распада синтезируется АТФ, гликолиз и гликогенолиз служит средством быстрого получения энергии в анаэробных условиях.
При переключении в аэробные условия от 1/5 до 1/6 общего количества молочной кислоты, возникшей при гликогенолизе, и, вероятно, вся молочная кислота, образовавшаяся при гликолизе, окисляются до СО2 и Н2О. От 4/5 до 5/6 общего количества молочной кислоты гликогенолитического происхождения идет на ресинтез гликогена путем обращения реакции гликогенолиза.
Энергия для этого черпается из реакций окисления, идущих в аэробных условиях.
Таким образом, в анаэробных условиях ПВК, образующаяся при дихотомическом распаде углеводов, становится акцептором гидрид-ионов (Н-) и протонов (Н+), снимаемых глицеральдегид-3-фосфатдегидрогеназой с 3-фосфоглицеринового альдегида. Регенерация окисленной формы НАД+ вследствие передачи гидрид-ионов на ПВК поддерживает течение гликолитического процесса. Последний неизбежно остановился бы, если бы все количество НАД+ оказалось насыщенным атомами водорода, ибо глицеральдегид-3-фосфатдегидрогеназа не смогла бы осуществлять свою функцию.
Полный набор ферментов гликогенолиза характерен для мышц и печени животных. Однако если в первых превалирует распад гликогена, то для второй более показателен его биосинтез.
У некоторых организмов, в частности в дрожжевых клетках, содержится мощная декарбоксилаза пировиноградной кислоты, способная в анаэробных условиях превращать ПВК в уксусный альдегид и СО2:

Начальная фаза реакции декарбоксилирования ПВК при участии дрожжевой пируватдекарбоксилазы (М = 185 000, состоит из двух субъединиц, каждая из которых несет молекулу тиаминпирофосфата в качестве кофермента и Mg2+ в качестве кофактора) рассмотрена ранее. Уксусный альдегид, образующийся при распаде оксиэтилтиаминпирофосфата (см. с. 161), восстанавливается за счет НАДН при участии другого фермента — алкогольдегидрогеназы, отличающейся тоже очень высокой активностью в дрожжевых клетках:
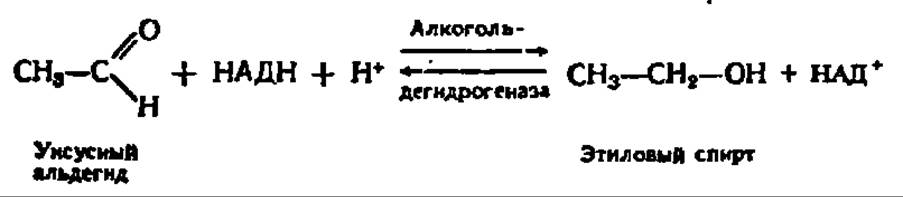
Механизм данной реакции, равно как и структура алкогольдегидрогеназы, также детально рассмотрен выше (см. рис. 53).
Так как конечным продуктом обмена углеводов в этом случае оказывается этиловый спирт, этот процесс называется спиртовым брожением. Как и при гликолизе, акцептирование атомов Н при брожении ацетальдегидом поддерживает течение реакции окисления 3-фосфоглицеринового альдегида, т. е. является условием осуществления процесса в целом.
Кроме спиртового брожения, у микроорганизмов существует еще ряд специфических путей утилизации трехуглеродных соединений, возникающих в результате дихотомического распада углеводов. Сюда относятся молочнокислое и пропионовокислое брожение, ацетоноэтиловое и ацетонобутиловое брожение, маслянокислое брожение и др.
В аэробных условиях ПВК окисляется. Реакция ускоряется мультиэнзимной системой, называемой пируватдегидрогеназным комплексом. Она идет в соответствии с уравнением
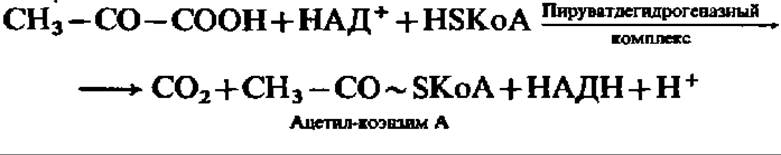
Характерно, что в результате реакции окисления ПВК в образующейся молекуле ацетил-КоА возникает макроэргическая связь. Она способствует его энергичному обмену в дальнейшем.
Структура пируватдегидрогеназного комплекса (ПДГК) и первая фаза ускоряемой при его посредстве реакции окислительного декарбоксилирования ПВК рассмотрены ранее (см. рис. 46 и уравнение реакции на с. 161). Как видно из этого уравнения, первая фаза процесса состоит в декарбоксилировании ПВК. Эта реакция ускоряется пируватдекарбоксилазой, которая входит в состав мультиэнзимного комплекса в количестве 12 димерных молекул (см. Е1 на рис. 46, Г); каждая из них несет две молекулы тиаминпирофосфата в качестве кофермента. Естественно, что оксиэтильный радикал, возникающий после декарбоксилирования ПВК, остается связанным с пируватдекарбоксилазой в виде оксиэтилтиаминпирофосфата (рис. 116, стадия 1).
Далее оксиэтильный радикал окисляется в ацетильный радикал, который переносится сначала на липоевую кислоту, а затем на коэнзим А. Оба эти процесса (окисление и перенос ацетильного радикала) ускоряются вторым компонентом пируватдегидрогеназного комплекса: липоат-ацетилтрансферазой (М = 70000). Она сосредоточена в центральной части комплекса в виде 24 молекул (см. Е1 на рис. 46, Г), упакованных, согласно современным данным, в виде куба, по 12 граням которого располагаются 12 димерных молекул пируватдекарбоксилазы, а по 6 плоскостям — 6 димерных молекул (М = 112 000) дигидролипоилдегидрогеназы (см. Е3 на рис. 46, Г). Общая молекулярная масса ПДГК кишечной палочки 4,6 млн. Да, а у высших организмов 7—8 млн. Да.
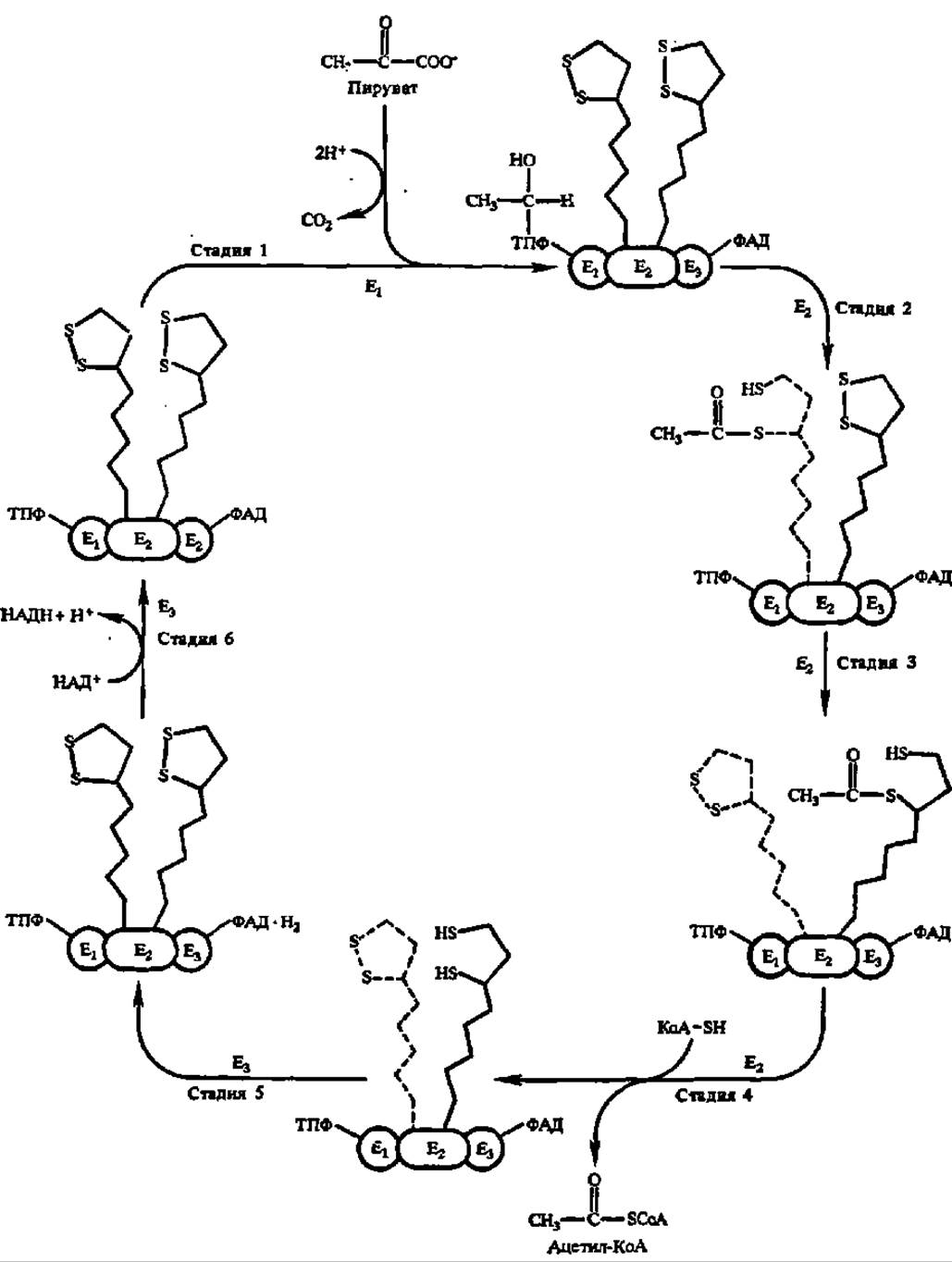
Рис. 116. Механизм действия пируватдегидрогеназного комплекса (пояснения в тексте)
Каждая молекула липоат-ацетилтрансферазы в качестве простетической группы содержит молекулу липоевой кислоты, соединенную с апоферментом через Е-аминогруппу радикала лизина. Такое присоединение липоевой кислоты обеспечивает ее подвижность в составе пируватдегидрогеназного комплекса (длина «ножки» — 1,5 нм) и беспрепятственный контакт как с пируватдекарбоксилазой, так и с дигидролипоилдегидрогеназой при условии использования не менее двух остатков липоевой кислоты (рис. 116, стадии 2, 3 и 4);
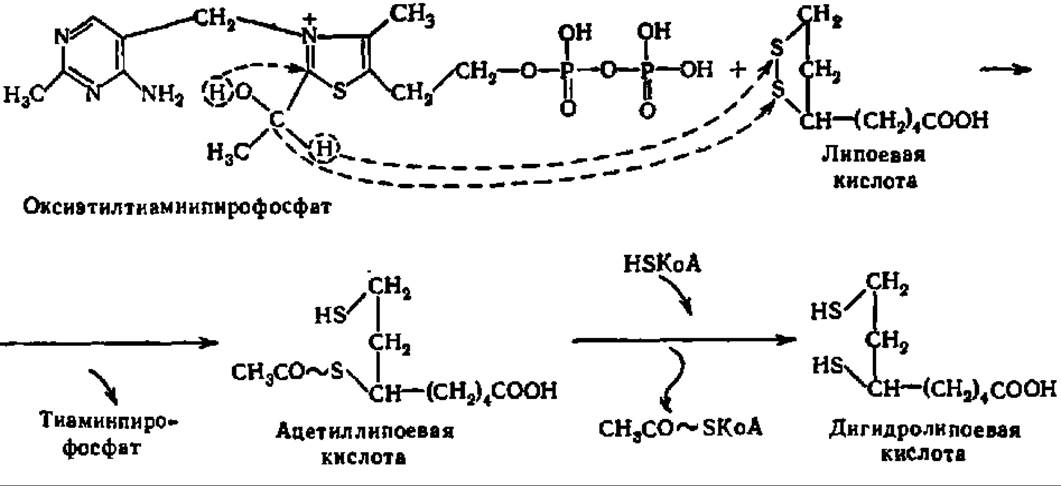
Окислительное декарбоксилирование ПВК завершается следующими (рис. 116, стадии 5 и 6) двумя реакциями. При посредстве третьего компонента мультиэнзимного комплекса, т. е. с помощью дигидролипоилдегидрогеназы (6 димерных молекул, содержащих по 2 молекулы ФАД в качестве кофермейта), дигидролипоевая кислота переходит в липоевую:

Поскольку коферментом дигидролипоилдегидрогеназы является ФАД, то конечно, именно он снимает непосредственно два атома Н с дигидролипоевой кислоты и передает их на НАД+. Поэтому в приведенном выше суммарном уравнении окислительного декарбоксилирования ПВК в качестве акцептора атомов Н выступает НАД+ (рис. 116). Пируватдегидрогеназный комплекс активен в дефосфорилированном состоянии: цАМФ-независимая протеинкиназа фосфорилирует Е1 и инактивирует его.
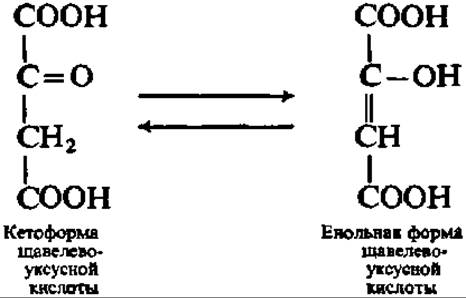
Ацетил-коэнзим А далее конденсируется со щавелевоуксусной кислотой (ЩУК), которая всегда есть в клеточном содержимом. Образуется лимонная кислота и освобождается коэнзим А. Каталитическую функцию в этой реакции выполняет конденсирующий фермент. Предполагают, что реакция идет в несколько стадий, которые могут быть выражены следующими уравнениями:
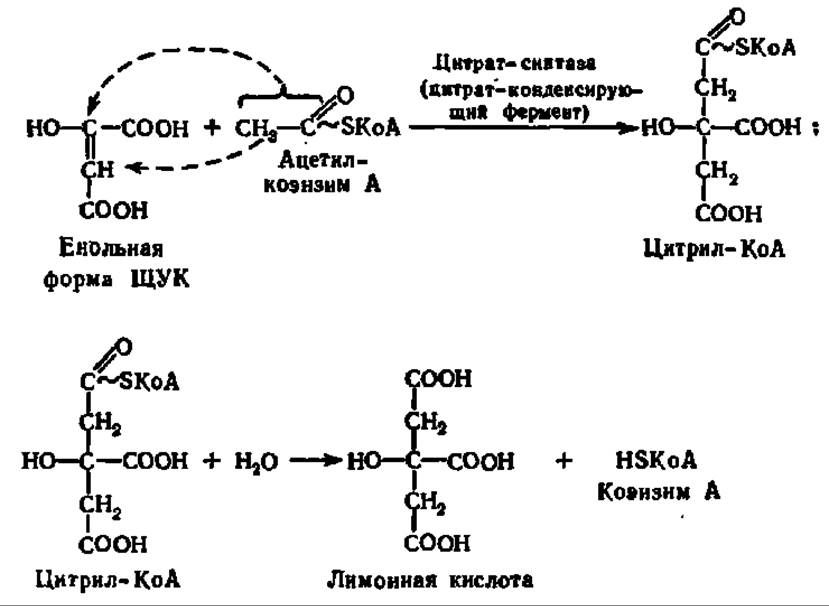
Образованием лимонной кислоты открывается специфический цикл химических реакций, приводящих к постепенному ее окислению до ЩУК, которая снова конденсируется с ацетил-коэнзимом А, так что образуется вновь лимонная кислота. По существу, следовательно, идет окисление ацетильных остатков до СО2 и Н2О. Этот цикл реакций получил название цикла трикарбоиовых и дикарбоиовых кислот, так как именно эти кислоты являются главными его компонентами (рис. 117). Его называют также циклом Кребса — по имени первооткрывателя, удостоенного за это Нобелевской премии в 1959 г. Таким образом, в конечном счете, ПВК окисляется до СО2 и Н2О.
Ферменты цикла трикарбоновых и дикарбоновых кислот (ЦТДК), ускоряющие единый метаболический многоступенчатый процесс окисления ацетильных гpyпп, возможно собраны в специфически построенный комплекс (метаболой), локализованный между расположенными друг против друга поверхностями внутренней мембраны митохондрий (рис. 118). В метаболоне, как полагают, осуществляется эстафетная передача промежуточных продуктов цикла от одного фермента к другому без их высвобождения в матрикс митохондрии. Поэтому процесс идет с большой скоростью. Радом с метаболоном ЦТДК располагаются пируватдегидрогеназный комплекс и, вероятно, метаболой ß-окисления высших жирных кислот, поставляющие ему СН3СО ~ SKoA.
Общая схема распада углеводов. Все сказанное выше о путях распада углеводов и о механизме реакций, осуществляющихся в процессе их деструкции, можно представить в виде следующей общей схемы (см. с. 357).
Из схемы видно, что глюкозо-6-фосфат занимает в этих процессах центральное место, а из промежуточных продуктов дальнейшего его распада узловые позиции принадлежат ПВК и рибулозо-5-фосфату.
Какая же роль в общем обмене углеводов организма отводится рассмотренным здесь путям распада углеводов: брожению, гликолизу и дыханию, апотомическому и дихотомическому, анаэробному и аэробному?
Зависимости между ними сложны и определяются как видовыми особенностями, так и условиями жизнедеятельности организмов. Например, объем гликолиза в тканях находится в прямой зависимости от поступления кислорода: последний подавляет процесс образования молочной кислоты (пастеровский эффект). Даже в различных тканях и органах одного и того же организма соотношения путей распада углеводов могут быть различными. Тем не менее можно установить и некоторые общие закономерности. Так, у подавляющего большинства организмов аэробный путь распада углеводов в общем превалирует над анаэробным, а дыхание подавляет гликолиз и брожение. Дихотомическому распаду углеводов принадлежит в целом более видное место, чем апотомическому. В значительной мере эти соотношения путей деструкции углеводов зависят от их энергетического эффекта. Последнее вполне естественно, так как одной из функций углеводов, в ряде случаев главной, является обеспечение организма энергией, выделяющейся при их анаэробном и аэробном распаде.
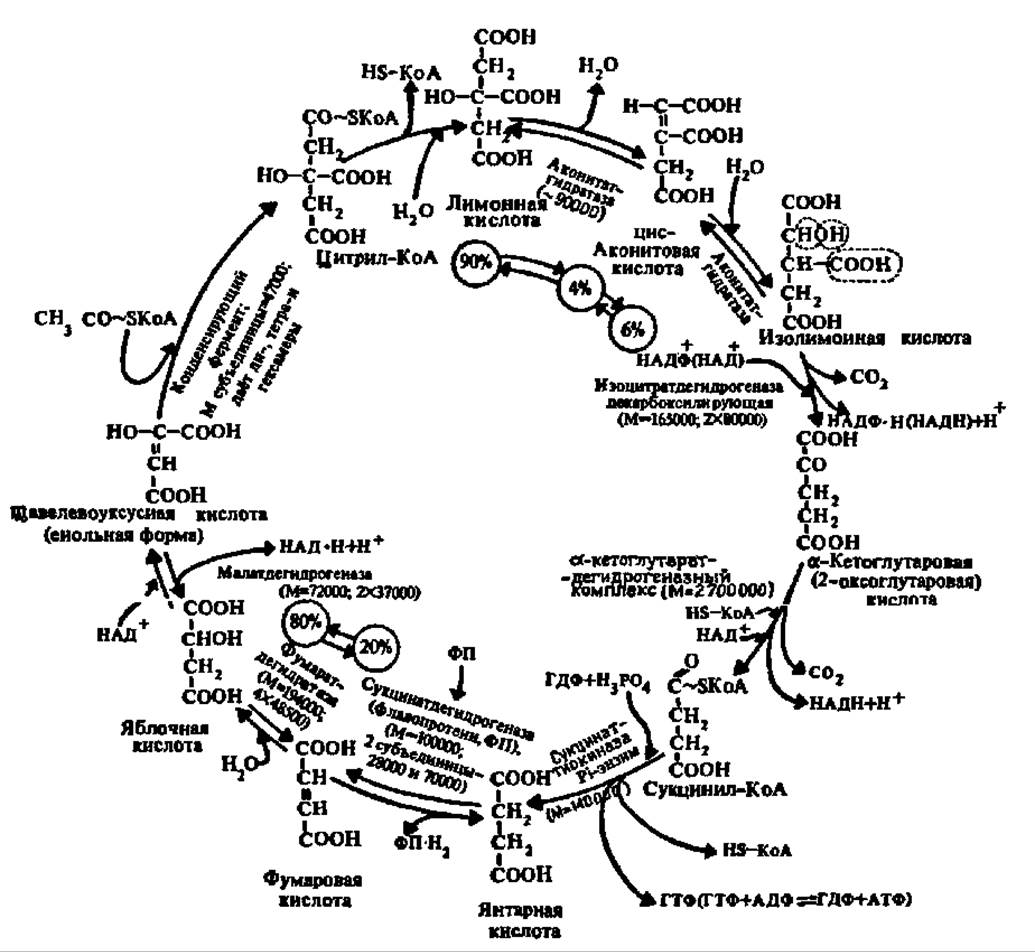
Рис. 117. Цикл трикарбоновых и дикарбоновых кислот (пояснения в тексте)
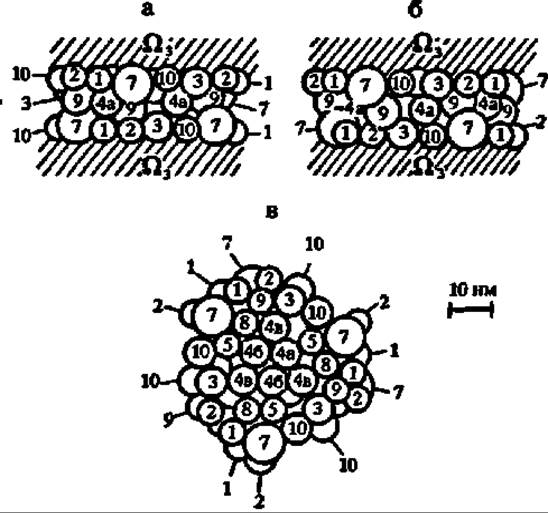
Рис. 118. Гипотетическая структура комплекса ферментов (метаболона) цикла трикарбоновых и дикарбоновых кислот (молекулярная масса метаболона ЦТДК ~ 8 млн Да; высота — 20 нм, диаметр — 50 нм):
а — вид прямо; 6 — вид сбоку; в — вид сверху; 1 — цитратсинтаза, 2 — аконитаза; 3 — изоцитратдегидрогеназа; 4а, 4б, 4в — а-кетоглутаратдегидрогеназный комплекс (а-кетоглутаратдегидрогеназа, транссукцинилаза и липоамиддегидрогеназа соответственно); 5 — сукцинаттиокиназа; 7 — фумараза; 8 — малатдегидрогеназа; 9 — аспартатаминотрансфераза; 10 — нуклеозиддифосфаткиназа; Ω — заякоривающие метаболой белки, включающие сукцинатдегидрогеназу
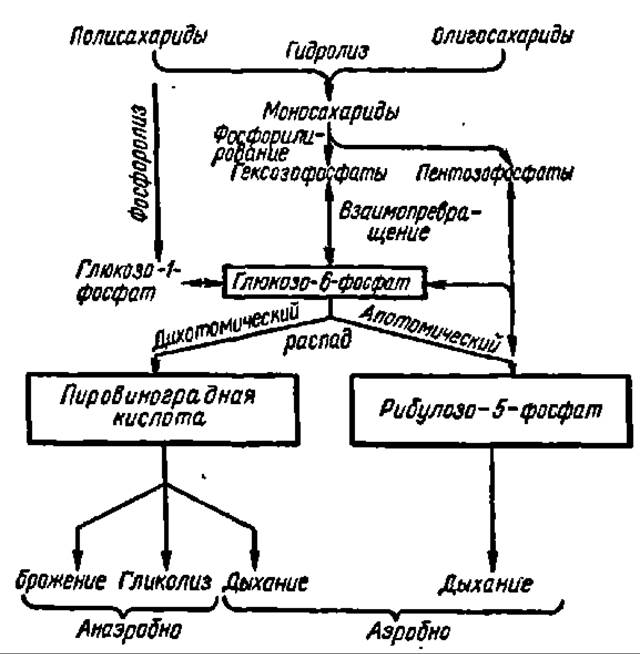
Схема 7. Пути распада углеводов (пояснение в тексте)
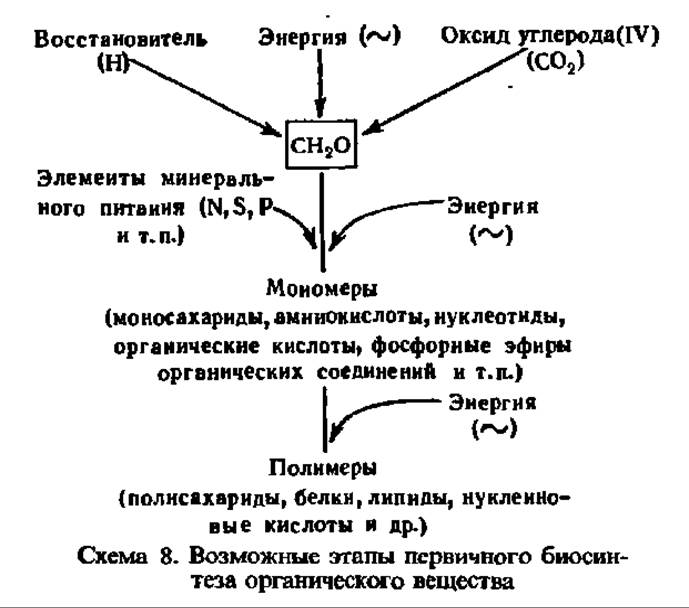
Синтез углеводов. Синтез простых углеводов. Простые углеводы возникают главным образом при первичном биосинтезе органического вещества на Земле. Этот процесс осуществляется автотрофными организмами — растениями, а также фотосинтезирующими и хемосинтезирующими бактериями. Первичный синтез органического вещества в природе идет путем восстановления СО2 атмосферы с одновременным формированием органических молекул, содержащих цепи углеродных атомов. В связях между атомами углерода и других элементов образующихся органических соединений заключена энергия, поэтому их новообразование сопровождается ее поглощением. В общем виде процесс первичного новообразования органического вещества принято изображать в виде следующей схемы (см. с. 358).
Гетеротрофные организмы, использующие для построения составных частей своего тела уже готовые органические вещества, не обладают способностью к первичному биосинтезу органических молекул, но могут образовывать их за счет перестройки органических соединений пищи. Естественно, что в числе новообразуемых гетеротрофами соединений находятся и простые углеводы; однако как исходные материалы для их построения, так и первичные источники энергии здесь принципиально отличаются от таковых у автотрофов.
Рассмотрим механизм первичного биосинтеза простых углеводов у автотрофных1 организмов. В простейшем случае у хемосинтезирующих бактерий источником энергии, которая трансформируется в стабильную энергию химических связей между атомами углерода, служат реакции окисления неорганических соединений, проходящие с выделением того или иного количества энергии (табл. 24).
Таблица 24 Энергетический эффект окислительных реакций у хемосинтезирующих бактерий
Уравнение реакции |
Количество энергии, выделявшейся на 1 г/моль окисленного вещества, кДж |
Бактерии |
Na2S2О3 + 21/2О2 + Н2→ Na2SО4 + H2SО4 |
882 |
Серобактерии |
S + 11/2О2 + Н2О → Н2SO4 |
493 |
|
NH3 + 1 1/2O2→ HNO2 + H2O |
276 |
Нитрифицирующие |
« |
бактерии |
|
H2 + 1/2O2 → H2O |
234 |
Водородные бактерии |
H2S + 1/2O2 → H2O + S |
171 |
Серобактерии |
2FeCO3 + 3H2O + 1/2O2 → 2Fe(OH)3 + 2CO2 |
167 |
Железобактерии |
HNO2 + 1/2O2 → HNO3 |
71 |
Нитрифицирующие бактерии |
Фотосинтезирующие бактерии и зеленые растения используют для первичного синтеза органических веществ энергию световых лучей, которая, например, для 6 X 1023 квантов красного света (в основном поглощаемого зелеными органами) равна примерно 167 кДж.
Хемосинтез, т. е. ассимиляция СO2 микроорганизмами за счет энергии, выделяемой при окислении неорганических соединений, впервые открыт в конце прошлого столетия С. Н. Виноградским.
1 Автотрофный — сам себя питающий, т. е. развивающийся на среде, свободной от других организмов и продуктов их жизнедеятельности.
Первичный акт, посредством которого энергия, освободившаяся при окислении неорганических соединений хемосинтезирующими бактериями или воспринятая фотосинтезирующими организмами, превращается в доступную для использования в химическом синтезе форму, состоит в трансформации этой энергии в энергию макроэргической связи АТФ. Иначе говоря, энергетическое обеспечение синтеза простых углеводов начинается с синтеза АТФ из АДФ и неорганического фосфата. Можно предполагать, что процесс хемосинтетического и фотосинтетического фосфорилирования идет, в общем, аналогично окислительному фосфорилированию (см. гл. X), т. е. перенос электронов при хемосинтезе и фотосинтезе вовлекает ряд энзиматических систем мембранного аппарата бактериальных и растительных клеток, результатом чего является возникновение мембранного потенциала—истинного двигателя реакции фосфорилирования аденозиндифосфорной кислоты: АДФ + Н3РO4 → АТФ + Н2O.
Открытие в составе сопрягающих мембран (см. гл. X) разнообразных организмов присутствия сопрягающих факторов схожей структуры, обеспечивающих протекание этой реакции, является доказательством единства путей акцептирования энергии в живых системах.
Одновременно с синтезом АТФ идет и вторая важнейшая для первичного биосинтеза органических веществ реакция—высвобождение атомов водорода, необходимых для восстановления СO2. Многие детали этого процесса неясны, однако установлено, что донором атомов Н в реакциях хемосинтетического и фотосинтетического восстановления С02 является в подавляющем большинстве случаев вода, а промежуточным акцептором их — НАДФ+, т. е. 2НАДФ+ + 2Н2O → 2НАДФН + 2Н+ + O2.
Восстановление СO2 непосредственно не идет. Оно осуществляется после связывания СO2 в результате реакции карбоксилирования уже достаточно сложного органического соединения — рибулозо-1,5-дифосфата, который образуется путем фосфорилирования рибулозо-5-фосфата — продукта апотомического распада глюкозы, всегда присутствующего в клеточном содержимом или возникающего из рибозо-5-фосфата при посредстве рибозофосфатизомеразы (М = 54000). Именно на этом этапе расходуется АТФ, необходимая для первичного биосинтеза углеводов:
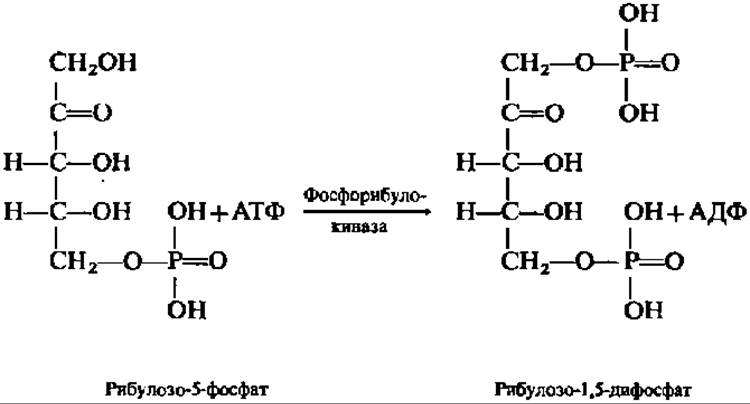
Фосфорибулокиназа, ускоряющая эту первую реакцию на пути акцептирования СО2, открыта А. Вейсбахом с сотрудниками еще в 1954 г., но свойства ее изучены лишь в последнее десятилетие; фермент широко представлен у фото- и хемоавтотрофов, высоко специфичен, абсолютно зависим от присутствия двухвалентных катионов, особенно Mg2+, обладает высокой молекулярной массой (240 000; тетрамер) и образует прочный комплекс с другими ферментами, участвующими в карбоксилировании рибулозо-1,5-дифосфата.
Механизм реакции карбоксилирования рибулозо-1,5-дифосфата достаточно ясен. Сначала это соединение, преобразуется в енольную форму:
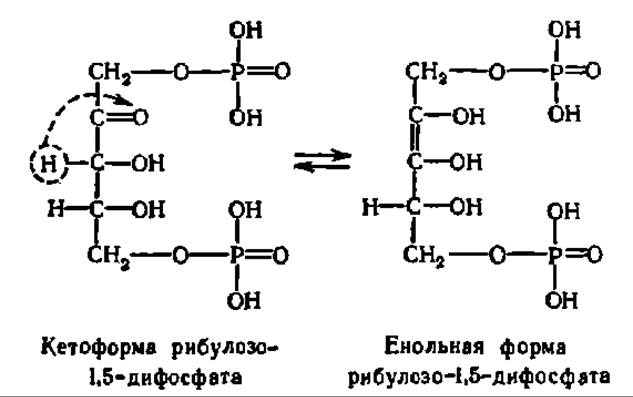
Она присоединяет СО2, и возникший промежуточный продукт расщепляется на две молекулы фосфоглицериновой кислоты:
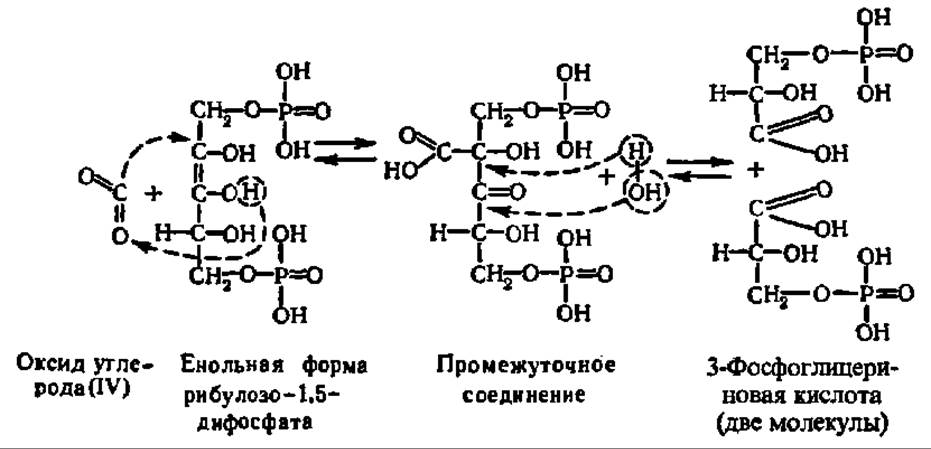
Приведенные выше уравнения представляют, конечно, лишь грубую схему тех тонких процессов, которые происходят во-время карбоксилирования рибулозо-1,5-дифосфата в активном центре специфического фермента, ускоряющего многостадийные преобразования при акцептировании СО2. Этот самый распространенный на Земле фермент называют рибулозодифосфаткарбоксилазой (РДФК). Его систематическое название — 3-фосфо-D-глицерат карбоксилиаза димеризующая. К настоящему времени он выделен из нескольких десятков объектов (высшие растения, зеленые водоросли, сине-зеленые водоросли, фототрофные и хемоавтотрофные бактерии). При молекулярной массе в 500 000—600 000 он обладает четвертичной структурой (8 больших субъединиц с М = 50000—60000 и 8 малых с М = 12000—15000), которая изучена методами рентгеноструктурного анализа и электронной микроскопии (рис. 119). Большие субъединицы являются каталитическими и несут центры связывания рибулозо-1,5-дифосфата и СО2, а малые — регуляторными и имеют ряд аллостерических центров для соединения с эффекторами (Mg2+, фруктозо-6-фосфат, HS — содержащие соединения и др.). Субъединицы РДФК синхронно синтезируются: большие — на рибосомах 70S в хлоропластах, малые — на рибосомах 805 в цитоплазме, спонтанно собираясь в нативный мультимер.
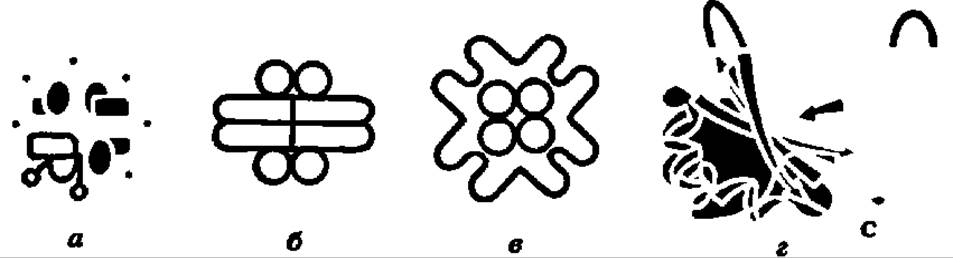
Рис. 119. Структура рибулозодифосфаткарбоксилазы:
четвертичная — высших растений (а) и водородных бактерий (б — вид сбоку, в — вид сверху), изученная электронно-микроскопически; третичная — малой субъединицы высших растений, изученная методом рентгеноструктурного анализа (г). Остальные пояснения — в тексте
В значительной мере выяснены структура и работа активного центра РДФК; предполагают, что рибулозо-1,5-дифосфат связывается в нем за счет взаимодействия фосфатных групп с радикалами лизина, енолизация субстрата идет при участии HS-групп (их насчитывается 96), акцептирование СО2 по енодиольной группе сопровождается переносом протонов при помощи гистидиновых остатков. РДФК активируется светом при помощи светочувствительного белкового фактора (М = 4000—8000) и ингибируется молекулярным кислородом, инициирующим оксигеназную (см. гл. X) функцию (т. е. окисление рибулозо-1,5-дифосфата в 3-фосфоглицериновую кислоту и 2-фосфогликолевую кислоту) этого уникального фермента, обеспечивающего фиксацию СО2 и первичный биосинтез углеводов в космических масштабах.
Как отмечено выше, центральную роль в осуществлении фотосинтеза играет трансформация энергии света в разность потенциалов мембраны фотосинтетического центра и сопряженный с этим синтез АТФ. Недавно, используя методы спектроскопии, рентгеноструктурного анализа и молекулярной генетики, удалось получить детальную картину событий, происходящих при фотосинтезе и выявить пространственное расположение и роль белков и пигментов, участвующих в этом процессе. За эту работу немецкие ученые Р. Хубер, И. Дайзенхофер и X. Михель удостоены Нобелевской премии 1988 г.
Возникшая в результате рассмотренных выше процессов фосфоглицериновая кислота восстанавливается в 3-фосфоглицериновый альдегид:
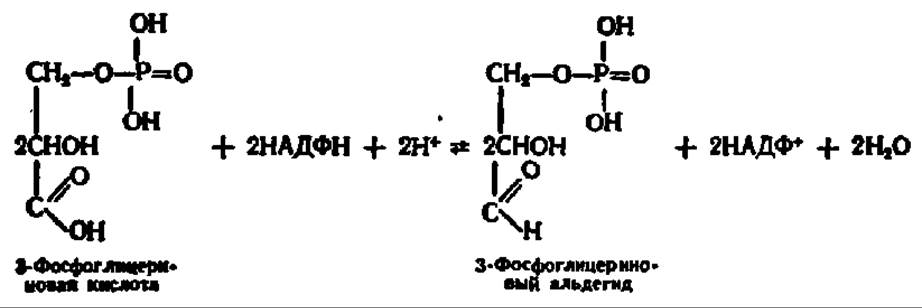
Данная реакция тоже идет более сложным путем, нежели показано в приведенном выше уравнении. Она представляет собой обращение процесса перехода 3-фосфоглицеринового альдегида в 3-фосфоглицериновую кислоту, осуществляющееся при дихотомическом распаде глюкозы. Напомним, что указанный процесс ускоряется глицеральдегид-3-фосфатдегидрогеназой и идет через 1,3-дифосфоглицериновую кислоту с тем лишь отличием, что содержащаяся в хлоропластах форма этого фермента специфична в большей степени к НАДФН, чем к НАДН, отличается очень высокой молекулярной массой (600 000) и специально приспособлена для восстановления фосфоглицериновой кислоты в фосфоглицериновый альдегид. Это значит, что при восстановлении 3-фосфоглицериновой кислоты в 3-фосфоглицериновый альдегид будет расходоваться АТФ (см. уравнение реакции на схеме 5, с. 344).
Следовательно, на этой стадии биосинтеза углеводов снова используется АТФ, образовавшаяся при фотосинтетическом или хемосинтетическом фосфорилировании и энергетически обеспечивающая новообразование углеводов. Здесь же, как следует из приведенного выше уравнения реакции восстановления 3-фосфоглицериновой кислоты, осуществляется в сущности восстановление акцептированного СО2 при посредстве НАДФН, возникшего одновременно с АТФ на первой фазе фотосинтетического процесса.
Дальнейший путь синтеза углеводов из 3-фосфоглицеринового альдегида, так же как и только что рассмотренная реакция восстановления 3-фосфоглицериновой кислоты, представляет обращение дихотомического пути распада углеводов: фосфоглицериновый альдегид переходит в фосфодиоксиацетон; при каталитическом воздействии альдолазы из упомянутых фосфотриоз синтезируется фруктозо-1,6-дифосфат, переходящий далее в глюкозо-6-фосфат. На этом этапе биосинтеза углеводов действует особый фермент — фруктозо-1,6-дифосфатаза (молекулярная масса фермента из растений и фото- и хемосинтезирующих организмов — 130000—190000; две субъединицы; отличается абсолютной специфичностью), обеспечивающая переход от фруктозо-1,6-дифосфата к фруктозо-6-фосфату, так как реакция:
фруктозо-6-фосфат+АТФ — фруктозо-1,6-дифосфат+А ДФ практически необратима. Фруктозо-6-фосфат, как известно, легко превращается в фосфорные эфиры других моносахаридов, что в итоге обеспечивает синтез всего набора природных моноз, а из них — дисахаридов и полисахаридов. Ход реакции первичного новообразования углеводов в процессе фотосинтеза дан на схеме 9.
Что касается синтеза простых углеводов организмами-гетеротрофами, то исходными веществами для этого могут служить продукты распада липидов, белков и других органических соединений. Центральным звеном в переходе этих соединений в простые (а далее и сложные) углеводы является образование ПВК, возникающей или непосредственно (например, при распаде аминокислот), или при посредстве глиоксилового цикла — при декарбоксилировании щавелевоуксусной кислоты (см. с. 395). Переход от ПВК к углеводам осуществляется путем обращения процесса дихотомического распада углеводов, следовательно, из каждых двух молекул ПВК образуется одна молекула фруктозо-1,6-дифосфата. Однако здесь есть одна особенность: переход от ПВК к фосфоенолпировиноградной кислоте идет обходным путем через щавелевоуксусную кислоту вследствие необратимости реакций превращения фосфоенолпирувата в ПВК. Так эта реакция протекает в печени, почках и других тканях животных, в листьях и корнях растений и у микроорганизмов. Только в мышцах действие пируваткиназы обратимо, и фосфоенолпировиноградная кислота возникает из ПВК и АТФ.
Кроме рибулозо-1,5-дифосфатного пути, существует еще два механизма акцептирования СО2, занимающих подчиненное положение по отношению к первому.
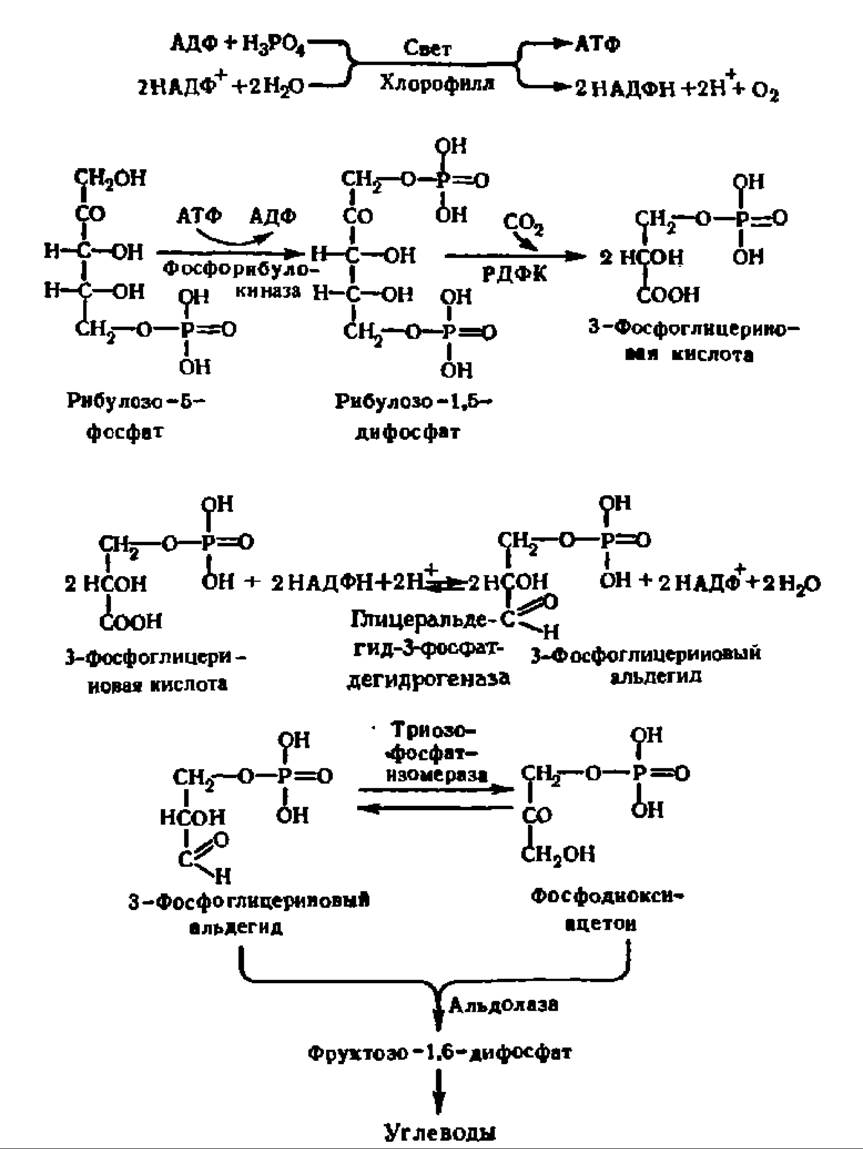
Схема 9. Механизм биосинтеза углеводов в растениях
Один из них состоит в карбоксилировании фосфоенолпировиноградной кислоты:
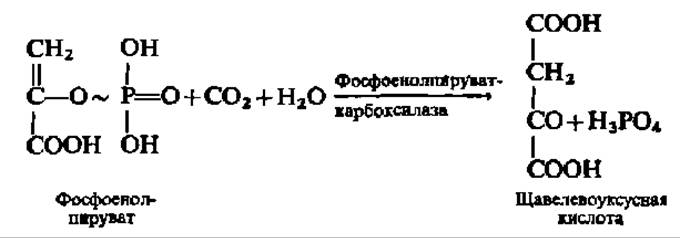
Фермент, ускоряющий эту реакцию, выделен из растений (обладающих С4-типом фотосинтеза), хемосинтезирующих и гетеротрофных бактерий (M ≈ 400000; четыре субъединицы).
Другой механизм акцептирования СО2 сводится к карбоксилированию ацильных производных коэнзима А (ацетил-КоА, пропионил КоА, сукцинил-КоА). Например:

Донорами атомов Н в указанных реакциях служат ферредоксины — железосодержащие белки негеминовой природы с М ≈ 6000 у бактерий и около 17000 в хлоропластах. Они содержат несколько атомов Fe, собранных в кластер и присоединенных, с одной стороны, к радикалам цистеина белковой части, а с другой — связанных с атомами лабильной серы.
Продукты, возникающие по фосфоенолпируват- и ацилкоэнзим-А-карбоксилазному механизму (пировиноградная, а-кетоглутаровая и щавелевоуксусная кислоты), используются для биосинтеза структурных элементов белков, углеводов, нуклеиновых кислот и липидов или обмениваются далее в цикле трикарбоновых и дикарбоновых кислот, усиливая, таким образом, метаболические возможности организма. Это послужило основанием для создания препарата-карбостимулина (NaHCО3 — 25 г, MgSО4 ∙ 7H2О — 3 г, MnSО4 ∙ 7H2О — 0,05 г, ZnSО4 ∙ 7H2О — 0,05 г, цитрат натрия — 7 г), находящего благодаря работам школы акад. М. Ф. Гулого все более широкое применение для повышения продуктивности животных, в медицинской практике и т.п.
Синтез олигосахаридов. Долгое время предпринимались безуспешные попытки доказать, что синтез олигосахаридов, в частности дисахаридов, представляет собой реакцию обращения их гидролиза. Однако, несмотря на многочисленные эксперименты такого рода, никто не сумел подобрать условия, при которых удалось бы направить вспять реакцию гидролиза, например сахарозы, протекающую при участии ß-фруктофуранозидазы (сахараза).
Оказалось, что биосинтез олигосахаридов осуществляется путем реакций трансгликозилирования. Перенос гликозильного остатка на один моносахарид идет с фосфорного эфира другого моносахарида и ускоряется специфической гликозилтрансферазой.
Исходными соединениями, с которых в процессе синтеза олигосахаридов гликозильный остаток энергично переносится на моносахарид, служат нуклеозиддифосфатсахара (НДФ-сахара).
Они были открыты Л. Лелуаром с сотр. (1950) и очень быстро привлекли к себе внимание как наиболее вероятные метаболиты в биосинтезе углеводов. В настоящее время известно свыше 50 представителей НДФ-сахаров.
Ясна и причина их особого значения в качестве доноров гликозильных остатков: при гидролизе НДФ-сахаров изменение уровня свободной энергии значительно выше, чем при гидролизе других доноров, а нуклеотидная часть их молекул способна обеспечить избирательность гликозилтрансферазной реакции.
В случае синтеза сахарозы, например, специфический фермент — сахарозосинтаза, ускоряет реакцию переноса остатка глюкозы с уридиндифосфатглюкозы на фруктозу (см. с. 365).
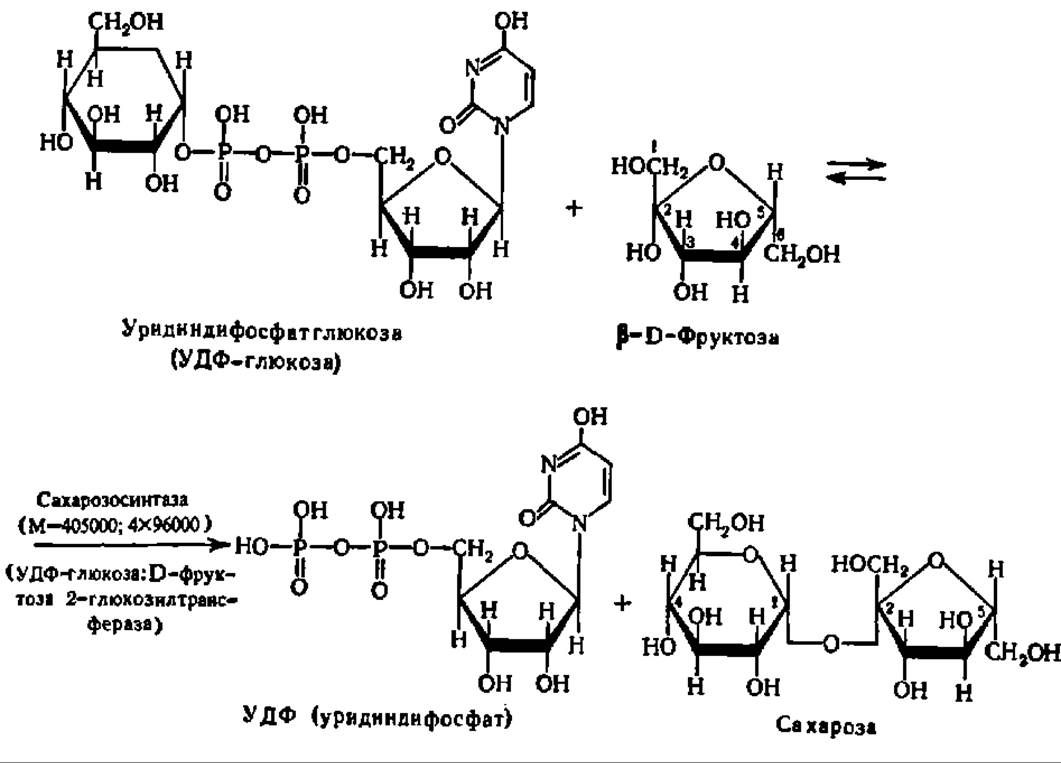
Долгое время считали, что именно так синтезируется сахароза в растениях (животные не синтезируют сахарозу, а лишь используют ее). Однако оказалось, что эта реакция ввиду ее обратимости служит для поддержания равновесия между сахарозой и УДФ-глюкозой и даже используется для наработки УДФ-глюкозы во время ее энергичного использования для биосинтеза крахмала и гликогена (см. следующий раздел этой главы). Этот процесс, в частности, идет в больших масштабах в клубнях растений, запасающих крахмал. Сахароза же в действительности синтезируется при помощи гликозилтрансферазной реакции из УДФ-глюкозы и фруктозо-6-фосфата:
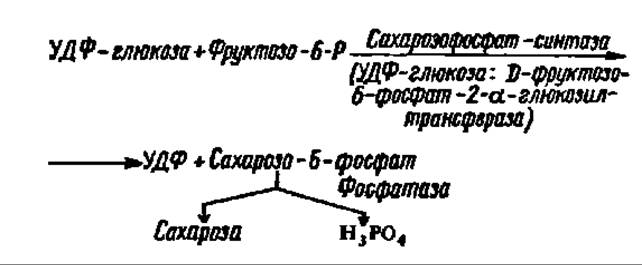
Как видно из уравнения реакции, сахарозофосфат-синтаза (молекулярная масса фермента из проростков пшеницы — 380000) действует в паре с фосфатазой, которая обеспечивает молниеносное отщепление остатка фосфорной кислоты от сахарозо-6-фосфата, чем полностью сдвигает реакцию вправо. Этот процесс идет в листьях растений, и образовавшаяся сахароза оттекает из них в клубни и корни. Аналогично синтезируется главный дисахарид насекомых, высших и низших грибов и микобактерий — трегалоза.
Будучи широко распространены в природе, НДФ-сахара синтезируются из фосфорных эфиров моносахаридов и соответствующих нуклеозидтрифосфатов путем нуклеотидилтрансферазных реакций, например:
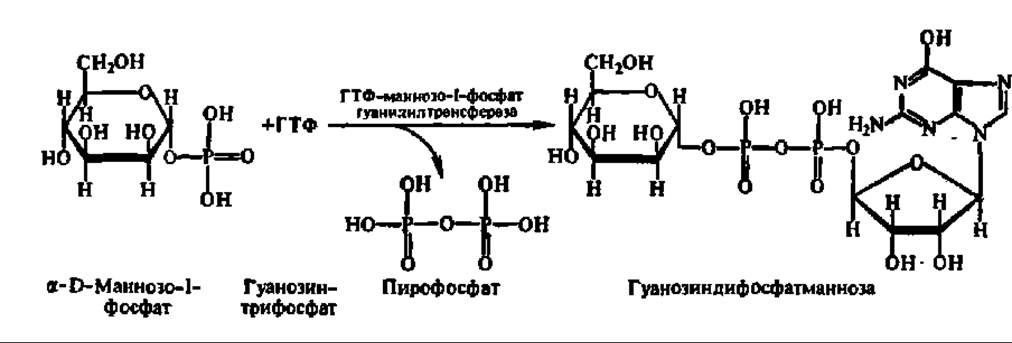
Известно более двух десятков нуклеотидилтрансфераз, ускоряющих реакцию переноса тех или иных нуклеотидных остатков на соответствующие фосфорные эфиры моносахаридов с освобождением пирофосфата. Наряду с фосфорными эфирами моносахаридов и нуклеозидцифосфатсахарами роль доноров гликозильных остатков в реакциях биосинтеза олигосахаридов могут выполнять сами олигосахариды, а также декстрины.
Из сказанного выше ясно, что биосинтез олигосахаридов идет путем переноса гликозильных остатков на моносахариды с разнообразных субстратов при участии в каждом конкретном случае соответствующих гликозилтрансфераз. При этом новообразование химических связей сопряжено с распадом их в тех соединениях, с которых идет перенос гликозильных остатков.
Синтез полисахаридов. Подобно синтезу олигосахаридов новообразование полисахаридов также идет путем трансгликозилирования. Полная аналогия существует и в характере субстратов, с которых переносятся гликозильные остатки на конец растущей цепи полисахарида. Ими могут быть фосфорные эфиры моноз, НДФ-сахара и олигосахариды. Реакции переноса остатков моносахаридов в процессе биосинтеза полисахаридов ускоряются соответствующими гликозилтрансферазами. Приведем несколько примеров.
Синтез амилозы, целлюлозы и подобных им 1,4-глюканов может происходить путем переноса гликозильных остатков с глюкозо-1-фосфата или аналогичных фосфорных эфиров моносахаридов. Эта реакция представляет собой обращение реакции фосфоролиза указанных соединений (см. с. 333). Однако более существенное значение имеет другой путь: биосинтез из НДФ-сахаров при участии соответствующих трансгликозидаз. Это уравнение реакции наращивания молекулы а-1,4-глюкана на один остаток глюкозы представлено на с. 367.
Как следует из приведенного уравнения реакции, перенос гликозильного остатка идет на невосстанавливающий конец молекулы синтезируемого полисахарида. Эта реакция может повторяться многократно, что обеспечивает ступенчатый синтез молекул полисахаридов, содержащих огромное число остатков моносахаридов. Характерная особенность реакций такого типа состоит в необходимости затравки, т. е. наличия в реакционной среде небольшого количества молекул полисахарида. Он как бы предопределяет тот тип связи, который возникает в процессе трансгликозилирования, и, следовательно, синтезируется полисахарид, одноименный с «затравочным». Роль затравки при биосинтезе некоторых разветвленных полисахаридов, таких, например, как частичковый гликоген, могут играть полипептидные цепи (см. рис. 104), содержащие олигосахаридные звенья.
Установлено, что при биосинтезе различных полисахаридов субстратами (а точнее, коферментами соответствующих трансгликозидаз) служат разные НДФ-сахара. Так, целлюлоза образуется с помощью гуанозиндифосфатглюкозы, полисахарид дрожжей (маннан) — с помощью гуанозиндифосфатманнозы и т. п. В связи с этим по-новому встает вопрос о специфичности реакции биосинтеза полисахаридов. Нуклеотидная часть НДФ-сахаров — это, по мнению Н. К. Кочеткова, «рукоятка», при помощи которой фермент располагает определенный сахар в нужном для осуществления реакции положении; она нужна также для «узнавания» гликозилтрансферазой соответствующего НДФ-сахара.
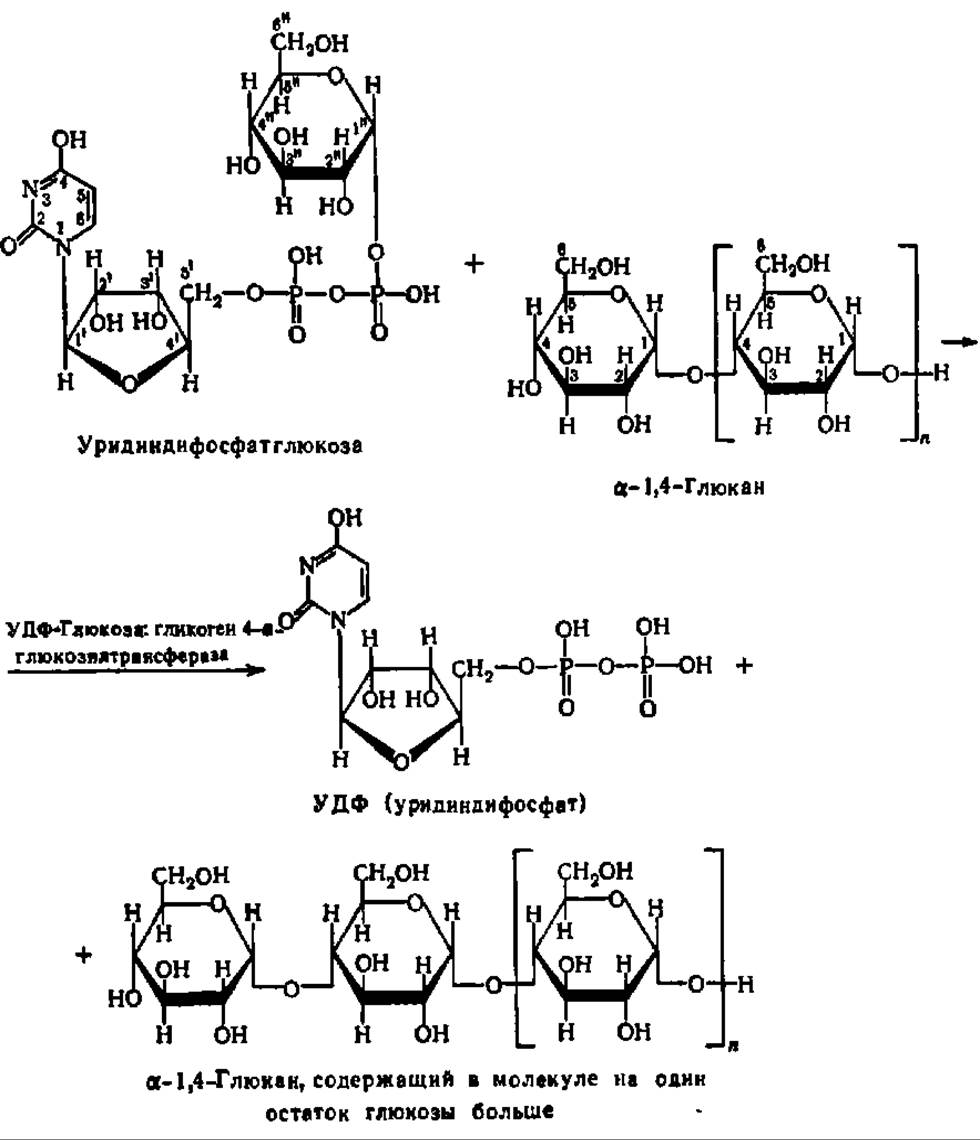
Конкретным представителем гликозилтрансфераз этого типа может служить гликогенсинтетаза. Она открыта Л. Лелуаром и сотр. (1957) в печени крысы, а сейчас обнаружена у многих животных и микроорганизмов. Гомогенный (без примеси прочно связанного с ним гликогена) фермент имеет М = 330—340 тыс. Да (4x85000), содержит около 23% а-спиралей и легко агрегирует. Соединяясь с гликогеном, образует комплексы с М = 5—6 млн., содержащие до 10 молекул фермента, локализованных по месту нередуцирующих концов наращиваемых олигосахаридных цепей.
Активность гликогенсинтетазы регулируется за счет реакций фосфорилирования (снижение) и дефосфорилирования (возрастание). Предполагают, что ее фосфорилирование осуществляется той же цАМФ-зависимой протеинкиназой, что и фосфорилирование гликогенфосфорилазы. Это подчеркивает весьма тонкие взаимоотношения систем, регулирующих синтез и распад гликогена.
Донором гликозильных остатков при синтезе полисахаридов могут служить и олигосахариды. Изучены реакции переноса остатков глюкозы на растущий конец цепи синтезируемого полисахарида с мальтозы, сахарозы и других олигосахаридов. Реакция аналогична описанной выше. Однако разнообразие типов связей, которые могут возникать при переносе гликозильных остатков с олигосахаридов на новообразуемый полисахарид, гораздо больше. Здесь перенос может идти не только на 4-й, но и на 6-й углеродный атом остатка моносахарида, что обеспечивает синтез 1,6-глюканов, а возможно, и полисахаридов разветвленного строения.
В последнее время внимание привлечено еще к одному источнику гликозильных и олигосахаридных остатков при биосинтезе полисахаридов и особенно полисахаридной части гликопротеинов и гликолипидов. Это — полиизопренилмонофосфат и дифосфатсахара, называемые также долихолфосфат и долихолдифосфатсахарами (от названия полиизопреноидного спирта — долихола, с числом изопреноидных остатков от 6 до 24). Особенно хорошо эти соединения представлены у микроорганизмов, хотя они, несомненно, принимают участие в новообразовании гликопротеинов у животных и растений:
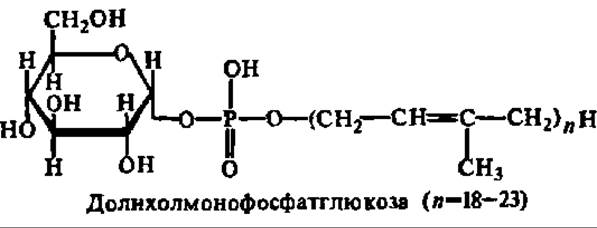
Встраиваясь полиизопреноидной частью в липофильную мембрану, они обеспечивают беспрепятственную переброску моносахаридных звеньев для достройки олигосахаридных фрагментов пептидогликанов и гликолипидов, происходящую при помощи соответствующих гликозилтрансфераз.
Особое значение реакции синтеза полисахаридов при участии долихолфосфатсахаров имеют для сборки пептидоглюканов клеточных стенок бактерий. Именно при их участии идет перенос углеводных фрагментов на пептидные группировки новообразуемого пептидоглюкана. Новым аспектом является участие долихолдифосфатолигосахаридов в N-гликозилировании белков при их посттрансляционной модификации.
Для реакций синтеза полисахаридов характерно также, что осуществляется перенос не только остатков моносахаридов, но и полигликозидных фрагментов с одного полисахарида на другой или же в пределах одной и той же молекулы. Реакции этого типа изучены у нас А. Н. Петровой и ею же впервые получен из скелетных мышц кролика гомогенный препарат соответствующего фермента (1970).
Примером может служить перенос части полиглкжозидной цепи у 1,4- глюкана из положения 4 в положение 6, катализируемый а-1,4-глюкан-ветвящим ферментом, систематическое название которого — а-1,4-глюкан: а-1,4-глюкан 6-а-(а-1,4-глюкано)-трансфераза. По данным А. Н. Петровой, этот фермент для осуществления реакции ветвления полисахарида нуждается в присутствии РНК с М ≈ 10000. Нуклеотидный состав и первичная структура этой РНК (31 н. о., из них 1/3 минорных) изучены, хотя механизм активирования ею фермента до конца не ясен. Мнение о том, что она является рибозимом, оказалось ошибочным.
Приводимая ниже упрощенная схема иллюстрирует действие а-1,4-глюканветвящего фермента:
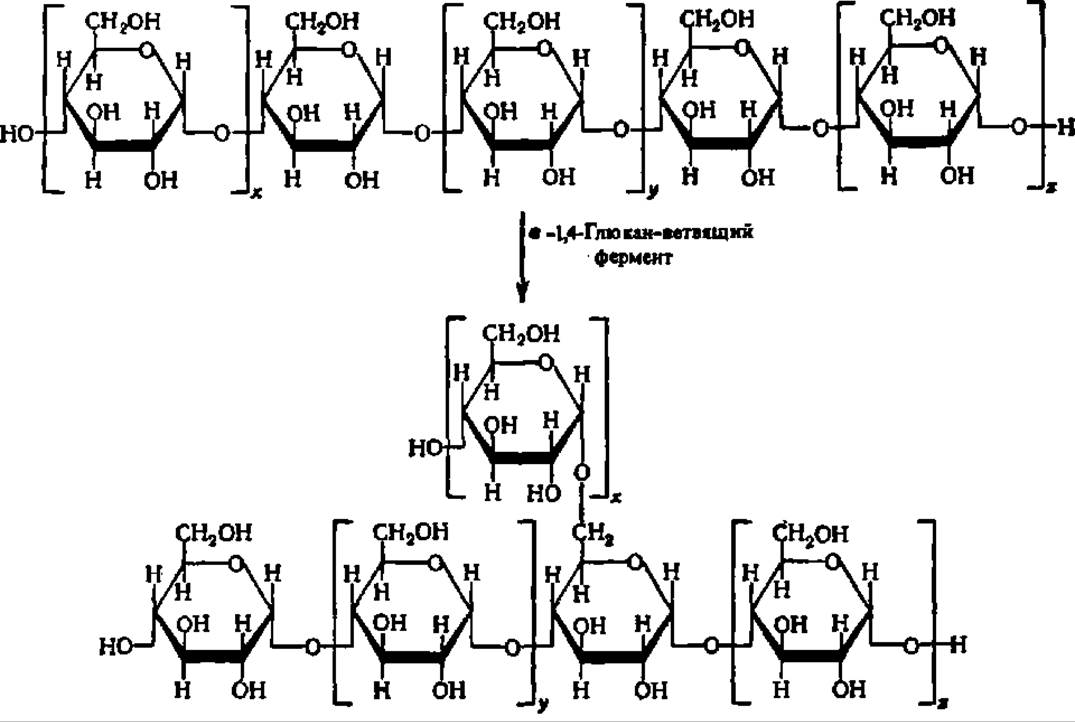
Так возникают в организме разветвленные молекулы полисахаридов (крахмала, гликогена и т. п.).
Использование промежуточных продуктов распада углеводов для синтеза других органических соединений. Выше было отмечено, что одна из функций углеводов в обмене веществ состоит в образовании продуктов распада, которые служат исходными веществами для синтеза многих других молекул. Из числа продуктов распада углеводов в этом смысле важны фосфоглицериновая кислота, фосфоенолпировиноградная кислота, пировиноградная кислота, ацетил-коэнзим А, эритрозо-4-фосфат, рибулозо-5-фосфат, а также партнеры цикла трикарбоновых и дикарбоновых кислот: щавелевоуксусная и а-кето-глутаровая кислоты. Они служат исходными соединениям для синтеза аминокислот, высших жирных кислот, глицерина, нуклеотидов и ряда других мономеров, используемых для построения белков, липидов, нуклеиновых кислот и других биополимеров. Конкретные примеры. превращений перечисленных выше соединений можно найти в предыдущих главах (см. разделы о синтезе аминокислот, пуриновых и пиримидиновых оснований и др.).