Основы биохимии - Филиппович Ю. Б. 1999
Биологическое окисление
История развития представлений о механизмах реакций биологического окисления
Разобраться в столь сложном явлении, как биологическое окисление, возможно лишь после ознакомления с теми концепциями, которые постепенно складывались по мере исследования этой проблемы.
Хотя более двухсот лет тому назад (1774—1777) А. Лавуазье считал дыхание очень медленным горением продуктов питания в организме, сходным со сгоранием угля, первая попытка выявить молекулярный механизм биологического окисления была предпринята лишь в следующем столетии Хр. Ф. Шёнбайном (1845—1868). Им была выдвинута идея о том, что необходимым условием протекания биологических окислительных процессов является активирование кислорода. Естественно, что конкретные пути этого активирования выглядели фантастически (предполагалось существование отрицательно-активной формы кислорода, тождественной озону и положительно-активной, названной антиозоном), но сама идея была плодотворна. Хр. Ф. Шёнбайн впервые высказал мысль о том, что биологическое окисление есть каталитический процесс. Ему же удалось экспериментально доказать образование Н2О2 при биологическом окислении.
На рубеже XIX и XX вв. наш соотечественник А. Н. Бах и независимо от него в Германии К. Энглер и В. Вилд выдвинули гипотезу об образовании пероксидов органических соединений как первом этапе биологического окисления. При этом молекула кислорода переводилась в активированное состояние за счет разрыва двойной связи в ней при посредстве «внутренней колебательной энергии» самого окисляемого соединения, обладающего кратной связью, и при участии ферментов — оксидаз в соответствии с такой, например, схемой:
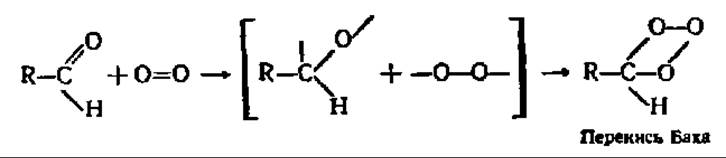
Возникшие пероксиды органических соединений, как и пероксид водорода, могут окислять другие вещества при каталитическом воздействии пероксидазы — фермента, достаточно в то время уже изученного:

Пероксид водорода может распадаться и иным путем, при участии фермента каталазы, о котором в этот период тоже накопилось довольно много сведений:
![]()
Вероятно поэтому, А. Н. Бах придавал большое значение участию в биологическом окислении пероксидазы и каталазы, полагая, что распределение Н2О2 между пероксидазой и каталазой может служить для регуляции этого процесса, например, в растениях.
В последующее время названным ферментам в окислительно-восстановительных процессах отводили более скромную роль.
Однако в свете данных об активных состояниях кислорода, о существовании супероксид-ионов и ферментов, принимающих участие в их обмене, — супероксиддисмутаз, значение, придаваемое Н2О2, каталазе и пероксидазе в реакциях биологического окисления, стало неуклонно возрастать.
Принципиально иной подход к расшифровке механизмов реакции биологического окисления был намечен в трудах В. И. Палладина, а вслед за ним — Г. Виланда. На основании опытов с дыхательными хромогенами (под ними он 3 подразумевал бесцветные вещества растительного происхождения, способные в присутствии оксидаз присоединять кислород и переходить при этом в пигменты, которые, в свою очередь, могли передавать присоединенный кислород окисляемому субстрату, одновременно обесцвечиваясь), В. И. Палладии впервые (1912) высказал идею о том, что биологическое окисление есть перенос водорода от окисляемого вещества навстречу кислороду с образованием воды в качестве конечного продукта. К этой идее В. И. Палладии пришел после того, как в одном из опытов обнаружил, что метиленовый синий, не содержащий в своем составе кислорода, может играть роль дыхательного хромогена, снимая атомы водорода с окисляемого субстрата:
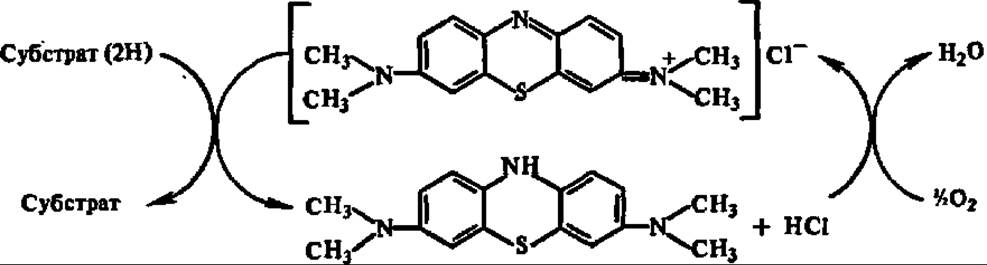
Стало ясно, что дыхательные хромогены являются не переносчиками кислорода, а акцепторами водорода.
Концепция В. И. Палладина довольно быстро получила подтверждение. Благодаря трудам Т. Тунберга, Д. Самнера, Г. Сомерса, В. Мак-Шена и других были выделены и охарактеризованы разнообразные дегидрогеназы, ускоряющие реакции окисления тех или иных субстратов при участии коферментов, являющихся акцепторами снимаемых с них атомов водорода.
Таким образом, в начале нашего столетия сложились две концепции биологического окисления: активирования кислорода и активирования водорода. Их противоборство продолжалось недолго: в 1925 г. Д. Кейлин в тканях ряда насекомых, а затем и в других аэробных биологических объектах открыл цитохромы — те недостающие ферменты, которые позволили несколькими годами позже связать активирование кислорода и водорода воедино. Этому способствовало обнаружение О. Варбургом (1928) цитохромоксидазы, получившей в то время наименование «дыхательного фермента Варбурга». Именно цитохромоксидаза оказалась тем ферментом, который непосредственно активирует кислород, а цитохромы — ферментами, снимающими электроны с водорода и передающими их цитохромоксидазе. Так впервые возникло представление об ансамблях ферментов дыхательной цепи, обеспечивающих реакции биологического окисления. В частности, оксидоредуктазная цепь, главной составной частью которой являются цитохромы, получила название цитохромной системы:
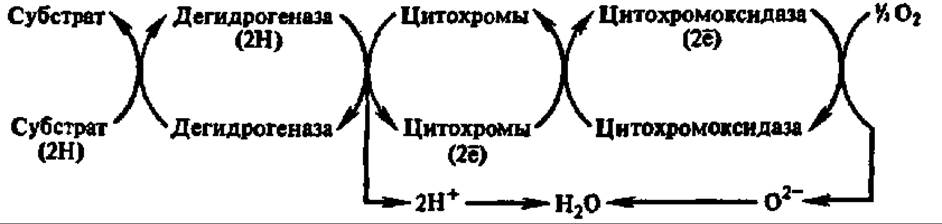
Ферменты, находящиеся в конце таких оксидоредуктазных цепей и непосредственно переносящие электроны на кислород, получили название терминальных оксидаз.
Позже (1947—1966) было показано, что цитохромной системой не исчерпывается перечень ферментных систем, способных активировать и водород, и кислород с последующим образованием из них молекул воды. Таких систем несколько. Простейшая из них наряду с пероксидазными реакциями — гликолатоксидазная:

Она представлена в растениях, у животных, грибов и бактерий. Гликолатоксидаза из листьев шпината — флавопротеин с М = 270 000 (8x37000). Четвертичная структура октамера гликолатоксидазы и третичная структура субъединиц недавно выяснена; имея около 10 нм в диаметре, октамер, составленный из 4 димеров, обладает полостью диаметром 6 нм. Гликолатоксидаза содержит флавинмононуклеотид (см. с. 120) в качестве кофермента; при его посредстве дегидрируется гликолевая кислота (активирование водорода). Вместе с тем гликолатоксидаза способна активировать кислород и передавать на него атомы водорода с восстановленного флавинмононуклеотида с образованием Н2О2, т. е. ей присуща флавопротеиноксидазная функция. Образовавшийся пероксид водорода распадается при участии каталазы.
В настоящее время изучено более двух десятков оксидаз флавопротеиновой природы, содержащих ФМН и ФАД в качестве коферментов (оксалатоксидаза, глюкозооксидаза, оксидазы L-аминокислот, ксантиноксидаза и др.).
Другой тип ферментных систем, обеспечивающих непосредственное окисление субстратов с передачей атомов водорода на кислород, представлен медьсодержащими оксидазами. Так как концентрированные растворы этих ферментов имеют синий цвет, их называют «синими оксидазами». Характерным представителем этой группы оксидаз является аскорбатоксидаза — белок с М = 130000—140000, содержащий 8 атомов Си на молекулу и состоящий из двух равных субъединиц. Она открыта А. Сцент-Дьердьи (1928) и очищена X. Таубером (1938). Уравнение реакции окисления аскорбиновой кислоты приведено на с. 171.
Развитие представлений об оксидоредуктазных системах, участвующих в осуществлении биологического окисления, сопровождалось уточнением их функций, классификации и номенклатуры входящих в их состав ферментов. Напомним (см. с. 117), что те дегидрогеназы, которые обеспечивают непосредственное дегидрирование субстратов, называются первичными. В отличие от них дегидрогеназы, получающие атомы Н от восстановленных коферментов первичных дегидрогеназ (НАДН, НАДФН, ФМН ∙ Н2, ФАД ∙ Н2 и др.) или от промежуточных акцепторов, на которые были переданы атомы водорода с первичных дегидрогеназ, отнесены к категории вторичных дегидрогеназ. Любые дегидрогеназы (и первичные, и вторичные), передающие атомы водорода на определенные акцепторы, называют редуктазами. Как отмечено ранее, все оксидоредуктазы, переносящие атомы водорода или электроны непосредственно на кислород, называют оксидазами.
В связи с этим было обращено внимание на окислительно-восстановительные системы, в которых между дегидрогеназами и молекулярным кислородом действует посредник. Атомы Н с восстановленной дегидрогеназы сначала поступают на окисленную молекулу посредника, а потом уже с нее — на О2. Обе реакции ускоряют специфическими ферментами. Конечным продуктом реакции является вода. Наиболее часто роль посредников играют хиноны и аскорбиновая кислота. При этом во взаимодействии с посредником участвует только восстановленный кофермент, например НАДН или НАДФН.
Первые данные о существовании совершенно нового пути биологического окисления, не похожего на все изученные ранее, были сообщены Е. Андре и К. Хоу (1932), открывшими в семенах сои особый фермент — липоксидазу. Он ускорял реакцию прямого присоединения атмосферного кислорода по двойным связям полиненасыщенных высших жирных кислот. Применение 18O2 и Н218О для экспериментального изучения аналогичных реакций позволило в 1955 г. одновременно и независимо Г. Мэзону с сотр. и О. Хайаиши с сотр. доказать наличие нового подкласса оксидоредуктаз — оксигеназ и изучить механизмы включения молекулярного кислорода при их посредстве в различные органические молекулы.
Согласно современным данным, липоксигеназа имеет молекулярную массу от 60000 до 190000 в зависимости от объекта выделения, распространена не только в растительном, но и в животном мире, существует в виде ряда молекулярных форм. В случае линолевой кислоты реакция идет в соответствии с уравнением:
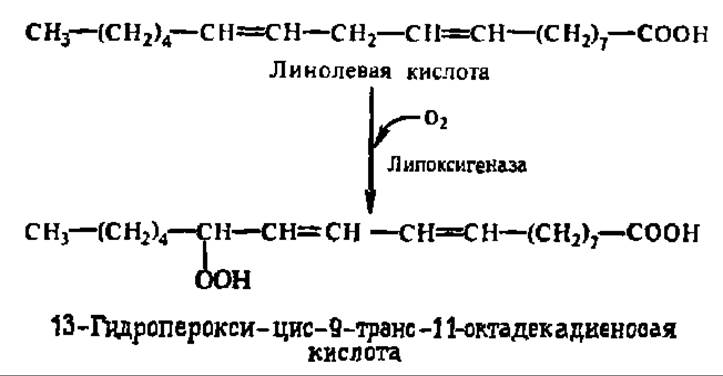
В случае фенолхиноновой системы схема процесса такова:
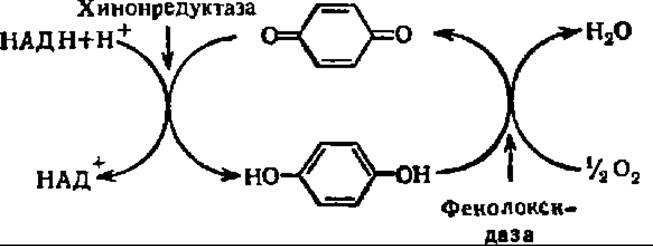
Липоксигеназы принимают участие в биосинтезе простагландинов, лейкотриенов и тромбоксанов из арахидоновой кислоты (см. с. 463).
Новую главу в учении о биологическом окислении составило открытие В. А. Энгельгардтом (1931) сопряжения реакций окисления органических соединений с фосфорилированием АДФ. Определяя содержание аденазинпирофосфорной кислоты, условно названной им пирофосфатом, в энергично дышащих эритроцитах голубя, он установил, что в атмосфере азота оно падает, а в атмосфере кислорода — возрастает до исходной величины. Повторение периода анаэробиоза с возвращением к аэробиозу сопровождалось новым циклом «распад — ресинтез» пирофосфата. Это означало, что существует какой-то механизм, при помощи которого энергия, выделяющаяся при окислении (в присутствии кислорода), не рассеивается, а используется для связывания, деминерализации неорганического фосфата, который обнаруживается в макроэргических связях аденозинтрнфосфата: АДФ + Н3РО4 → АТФ + Н2О.
Позже идеи В. А. Энгельгардта были более детально разработаны В. А. Белицером с сотр., внесшими существенный вклад в развитие проблемы сопряжения окисления с фосфорилированием в период ее становления. В частности, В. А. Белицером было введено понятие: коэффициент окислительного фосфорилирования, т. е. отношение количества молей синтезированной АТФ к количеству молей кислорода, использование которого обеспечивало этот синтез. Будучи вычислено после ряда измерений, значение коэффициента оказалось в среднем близким к 3,0, что свидетельствовало о том, что перенос двух атомов водорода (а в дальнейшем — двух электронов) по дыхательной цепи ферментов сопровождается синтезом трех молекул АТФ, т. е. существует три пункта сопряжения окисления с фосфорилированием АДФ.
Исследование тонких механизмов сопряжения окисления с фосфорилированием, являющееся фундаментом биоэнергетики, продолжается уже более полустолетия и еще далеко от завершения. Ниже будет освещено его нынешнее состояние, отражающее всю сложность современных проблем биологического окисления.