Основы биохимии Том 2 - А. Ленинджер 1985
Биоэнергетика и метаболизм
Окислительное расщепление аминокислот. Цикл мочевины
Наследственные нарушения катаболизма фенилаланина
У человека известно много различных наследственных нарушений аминокислотного обмена. В основе всех этих нарушений (большинство из них встречается редко) лежит мутация какого-нибудь гена, кодирующего определенный фермент, участвующий в превращениях данной аминокислоты. Под контролем мутантного гена синтезируется дефектный фермент, у которого в том или ином ключевом участке полипептидной цепи может стоять «неправильная» аминокислота; кроме того, какой-нибудь аминокислотный остаток может быть утрачен или, наоборот, включен в полипептидную цепь. В одних случаях такой наследственно измененный фермент неактивен вообще, а в других проявляет лишь часть присущей ему активности, поскольку характерное для него значение КМ (или Vmax) не соответствует норме. Большинство врожденных нарушений аминокислотного обмена у человека сопряжено с накоплением тех или иных промежуточных продуктов этого обмена. При некоторых наследственных заболеваниях такого рода нарушается нормальное развитие нервной ткани, что приводит к умственной отсталости.
Фенилаланин - тирозиновый путь заслуживает в этом смысле специального упоминания, поскольку три ферментативных этапа этого пути особенно уязвимы, т.е. подвержены генетическим изменениям, в результате которых возникают три вида врожденных нарушений обмена. У некоторых людей дефект затрагивает первый фермент данного метаболического пути (рис. 19-9) - фенилаланин—4-монооксигеназу (ее называют также фенилаланин-гидроксилазой), катализирующую гидроксилирование фенилаланина до тирозина. Этот дефект служит причиной заболевания, которое носит название фенилкетонурии. Фенилаланин-монооксигеназа катализирует реакцию, в которой один из двух атомов молекулы кислорода O2 включается в фенилаланин, чтобы образовать гидроксильную группу тирозина; второй атом кислорода восстанавливается при этом до Н2O за счет NADH, который также требуется для этой реакции:
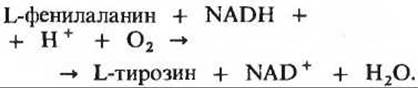
При наследственном дефекте, затрагивающем фенилаланин-4-монооксигеназу, на первый план выступает второстепенный путь обмена фенилаланина, в норме мало используемый. На этом второстепенном пути фенилаланин претерпевает трансаминирование в реакции с a-кетоглутаратом, что приводит к образованию фенилпирувата (рис. 19-10):
![]()
Однако дальнейшим превращениям фенилпируват не подвергается, т.е. это тупиковый путь; фенилпируват (а также и фенилаланин) накапливается в крови и тканях, а затем выводится с мочой. Избыток фенилпирувата в крови у новорожденного нарушает нормальное развитие мозга и служит причиной умственной отсталости. Фенилкетонурия (ФКУ) - одно из первых врожденных нарушений обмена, открытых у человека. При достаточно раннем выявлении фенилкетонурии можно с помощью соответствующей диеты создать условия для нормального развития и избежать умственной отсталости. Из рациона должны быть при этом исключены любые продукты, в состав которых входят белки с высоким содержанием фенилаланина. Поскольку почти во всех белках содержится какое-то количество фенилаланина и так как в малых количествах он все же необходим для нормального роста (это одна из незаменимых аминокислот; гл. 26), состав такого рациона должен контролироваться очень тщательно. Природные белки, например казеин молока, следует предварительно подвергать гидролизу и удалять из них фенилаланин.
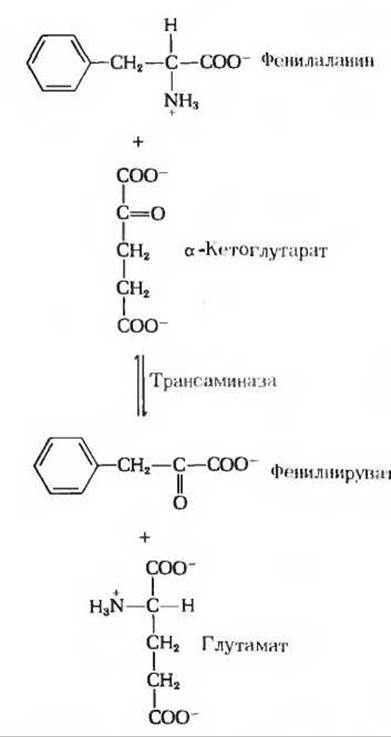
Рис. 19-10. Образование фенилпирувата на альтернативном пути, действующем при фенилкетонурии.
Выявить фенилкетонурию и назначить ребенку соответствующую диету необходимо в первые же недели после рождения, в противном случае неизбежна необратимая задержка умственного развития. При отсутствии лечения многие больные фенилкетонурией не доживают до 25 лет; других приходится всю жизнь содержать в соответствующих учреждениях и тратить на это много труда и средств (дополнение 19-2). Фенилкетонурия - серьезная проблема здравоохранения. Болезнь эта достаточно широко распространена: на 10000 новорожденных приходится в среднем один с таким дефектом. В США в большей части штатов все новорожденные подвергаются обязательной проверке на фенилкетонурию. Обнаружение этого заболевания не представляет затруднений: требуется только определить содержание фенилаланина и фенилпирувата в моче.
В некоторых случаях в результате генетической мутации дефектным оказывается также четвертый фермент фенилаланинового пути (рис. 19-9)-гомогентизат—1,2-диоксигєназа. У людей с таким генетическим дефектом не подвергается дальнейшему расщеплению один из промежуточных продуктов катаболизма фенилаланина-гомогентизат, который накапливается в жидкостях тела и выводится из организма с мочой. На воздухе такая моча темнеет. Объясняется это тем, что из-за разложения части мочевины с образованием аммиака pH мочи сдвигается в щелочную сторону; гомогентизат при этом спонтанно окисляется атмосферным О2 и превращается в темный пигмент, сходный с тем, который содержится в коже людей черной расы. Это врожденное нарушение обмена носит название алкаптонурии. У носителей такого дефекта здоровье явным образом ни в чем не страдает, если не считать беспокойства, вызываемого у них самим видом черной мочи. Это тоже немаловажно: известны случаи, когда люди действительно заболевали от одного только страха - черная моча считалась дурным знаком.
Дополнение 19-2. Значение некоторых наследственных болезней для человека, общества и экономики
В настоящее время у человека известно по приблизительным подсчетам уже свыше 2000 различных наследственных нарушений или болезней и число их быстро растет. Ежегодно в США рождается более 120 000 детей с наследственными заболеваниями. Во многих случаях это тяжкое несчастье отдельных людей, всю глубину которого не всегда можно себе даже представить. Тяжелым бременем ложатся эти болезни и на общество; необходимо, следовательно, позаботиться о том, чтобы будущие родители имели возможность вовремя получить нужные рекомендации и чтобы соответствующие знания шире распространялись в обществе.
Фенилкетонурия может служить примером довольно часто встречающейся генетической болезни, которую легко распознать и которая поддается лечению. Если выявить это патологическое состояние сразу же после рождения ребенка и в течение первых 6 лет его жизни очень строго соблюдать определенную диету, то он вырастет нормальным человеком. Обнаружение и лечение болезни стоят весьма недешево. Но отказ от них обходится в конечном счете намного дороже, даже если оставить в стороне чисто гуманные соображения. В 1980 г. стоимость теста на ФКУ составляла около двух долларов на одного ребенка. Это означает, что на 3 млн. детей, ежегодно рождающихся в США, необходимо затрачивать около 6 млн. долларов. Поскольку частота положительных тестов на ФКУ равна 1 на 10 000 новорожденных, такое тестирование должно выявлять ежегодно 300 детей, нуждающихся в специальной диете (без фенилаланина). Затраты на подобную диету превышают 1000 долларов в год на одного ребенка. Таким образом, ежегодно на лечебное питание для этих детей (вплоть до 6-летнего возраста) придется выделять 1,8 млн. долларов. В целом по стране стоимость этой программы будет приближаться к 8 млн. долларов в год, и цифра эта, очевидно, будет расти. Эта сумма, которую потребуется ежегодно расходовать всего на 300 детей, может показаться огромной, но мы сейчас увидим, что альтернативный путь связан с гораздо большими затратами. Если у новорожденных не будут проводиться тесты на ФКУ, эти 300 детей будут, вероятно, обречены на то, чтобы провести свою жизнь (в среднем около 30 лет) в специальных учреждениях для умственно отсталых, где общая стоимость содержания одного человека составляет 10 000 долларов в год. Отсюда следует, что, затрачивая на выявление и лечение фенилкетонурии в целом по стране свыше 8 млн. долларов в год, мы в конечном счете сберегаем вдесятеро большую сумму. Таким образом, по крайней мере в отношении фенилкетонурии расходы вполне оправдываются. По всей вероятности, так же обстоит дело и с другими наследственными болезнями.
При некоторых генетических заболеваниях проверка будущих родителей позволяет выявить носителей дефектных генов. Такая проверка не гарантирует от ошибок, и в большинстве случаев ее проводят на добровольной основе. Удается, например, выявить носителей серповидноклеточной анемии (разд. 8.17) или болезни Тея-Сакса (гл. 21). К сожалению, для многих других генетических болезней сделать это невозможно. В некоторых случаях метод амниоцентеза позволяет обнаружить ту или иную патологию еще у плода. Так может быть выявлена, в частности, болезнь Тея-Сакса (гл. 21). Правда, для ряда генетических болезней, выявляемых методом амниоцентеза, единственным возможным «лечением» является аборт, а это ставит людей перед трудным выбором.
Всего страшнее те генетические болезни, которые мы не в состоянии предсказать на основе проверки будущих родителей, не можем выявить достаточно рано или пока еще совершенно не умеем лечить. Жертвы таких болезней требуют ухода, затраты на который обычно не под силу частным лицам, так что обществу приходится брать на себя заботу об этих людях. Если бы даже биохимики и могли для каждой генетической болезни указать причину в виде дефектной структуры гена, то все равно одной биологии недостаточно для решения тех социальных и этических проблем, которые порождаются этими болезнями.
В табл. 19-1 указаны и некоторые другие врожденные нарушения аминокислотного обмена.
Таблица 19-1. Некоторые врожденные нарушения аминокислотного обмена у человека
|
Нарушение |
Затронутый фермент |
|
Альбинизм |
Тирозин—3-монооксигеназа |
|
Алкаптонурия |
Гомогентизат—1,2-диоксигеназа |
|
Аргининосукцинатацидемия |
Аргининосукцинатлиаза |
|
Гомоцистинурия |
Цистатионин—ß-синтаза |
|
Болезнь кленового сиропа (лейциноз) |
Дегидрогеназа а-кетокислот с разветвленной цепью |
|
Фенилкетонурия |
Фенилаланин—4-монооксигеназа |
|
Гипервалинемия |
Валин-трансаминаза |