Биохимия человека Том 1 - Марри Р. 1993
Метаболизм белков и аминокислот
Порфирины и желчные пигменты
Порфирии
Порфириями называют гетерогенную группу заболеваний, характеризующихся повышенным выделением порфиринов или их предшественников. Некоторые формы порфирии являются наследственными, другие — приобретенными. Было предложено несколько различных классификаций порфирий. Наследственные формы удобно разделить на три большие группы — эритропоэтические, печеночные и такие формы, при которых нарушения метаболизма наблюдаются одновременно в эритропоэтической и печеночной тканях (табл. 33.2). Для большинства наследуемых форм характерно наличие метаболических нарушений во всех тканях, однако проявляются они по каким-то причинам в каком-то одном типе тканей. Ниже приводится описание биохимических нарушений, характерных для порфирий.
Таблица 33.2. Классификация порфирий человека 1)
|
Состояние |
Тип наследования |
Установленный или предполагаемый дефектный фермент |
Ткань, в которой выражены метаболические нарушения |
|
Врожденная эритропоэтическая порфирия (болезнь Гюнтера) Печеночные формы порфирии |
Аутосомно-рецессивный |
Уропорфириноген-І-синтаза и(или) уропорфириноген-ІІІ-косинтаза |
Эритроидные клетки |
|
Перемежающаяся острая порфирия |
Аутосомно-доминантный |
Уропорфириноген-І-синтаза |
Печень |
|
Наследственная копропорфирия |
Аутосомно-доминантный |
Копропорфириногеноксидаза |
Печень |
|
Мозаичная порфирия |
Аутосомно-доминантный |
Протопорфириногеноксидаза |
Печень |
|
Поздняя порфирия кожи |
Аутосомно-доминантный (?) |
Уропорфириноген-декарбоксилаза |
Печень |
|
Токсическая порфирия |
Приобретенное состояние |
Различные нарушения |
Печень |
|
Протопорфирия |
Аутосомно-доминантный |
Феррохелатаза |
Эритроидные клетки и печень (?) |
1) Воспроизведено, с разрешения, из обзора Meyes U. A., Schmid R. The porphyrias. In: The Metabolic Basis of Inherited Disease, 4th ed. Stanbury J. B., Wyngaarden J. B., Fredrickson D. S. (eds). McGraw-Hill, 1978.
Для каждого типа порфирии характерен набор экскретируемых с мочой порфиринов и их предшественников. Эти данные и их связь с различными стадиями синтеза гема приведены на рис. 33.11.
Перемежающаяся острая порфирия (ПОП) является признаком аутосомно-доминантной наследственной болезни человека, которая обычно проявляется только после достижения половой зрелости. Ее причиной является наследуемая частичная недостаточность уропорфиряноген-І-синтазы. Больные гетерозиготны по дефектному структурному гену, поэтому активность уропорфириноген-І-синтазы в их клетках составляет 50% от нормы. Пациенты с ПОП экскретируют с мочой большие количества порфобилиногена и АЛК. Оба этих соединения бесцветны, но порфобилиноген на свету и воздухе самопроизвольно образует два окрашенных продукта — порфобилин и порфирии. Это является причиной потемнення мочи при ее стоянии на свету при доступе воздуха.
Порфобилиноген и АЛК присутствуют в плазме и спинномозговой жидкости больных, особенно в период резкого обострения. Лекарственные препараты и стероидные гормоны, метаболизм которых требует участия гемсодержащих белков, таких, как цитохром Р-450, могут ускорять наступление обострения. Соединения, индуцирующие порфирию в процессе метаболизма, повышают потребление гемовых белков и тем самым снижают внутриклеточную концентрацию гема; это приводит к дерепрессии синтеза АЛК-синтазы. Повышение активности АЛК-синтазы и частичное блокирование уропорфириноген-І-синтазы приводит к значительному накоплению АЛК и порфобилиногена; это сопровождается острыми болями в животе, рвотой, запором, сердечно-сосудистыми нарушениями, а также нервно-психическими расстройствами. Следует отметить, что, согласно экспериментальным данным, снижение содержания гема угнетает активность триптофанпирролазы и приводит к накоплению нейроактивных соединений — триптофана и 5- гидрокситриптамина.
У пациентов с ПОП не наблюдается повышенной чувствительности к свету, которая характерна для других печеночных порфирий. Этого следовало ожидать, поскольку у больных не накапливаются ни порфирины, ни порфириногены, так как метаболическое нарушение синтеза гема происходит на стадии, предшествующей образованию первого порфириногена (уропорфириногена).
Как отмечено выше, метаболические нарушения обнаруживаются и в других клетках, в частности, в эритроцитах, а также в выращенных в культуре фибробластах или клетках амниотической жидкости; однако повышенная активность АЛК-синтазы, приводящая к перепроизводству АЛК и порфобилиногена, наблюдается преимущественно в печени. Видимо, это связано с тем, что печень является тем самым органом, в котором протекает метаболизм индуцирующих агентов. Острая порфирия служит одним из редких примеров болезни, фенотипически выраженной у гетерозигот, при этом недостаточность фермента составляет только 50%.
Как и можно было предсказать на основании предложенного механизма регуляции синтеза АЛК-синтазы системой репрессии-дерепрессии, введение гематине пациентам с ПОП может уменьшить индукцию АЛК-синтазы и тем самым облегчить протекание заболевания.
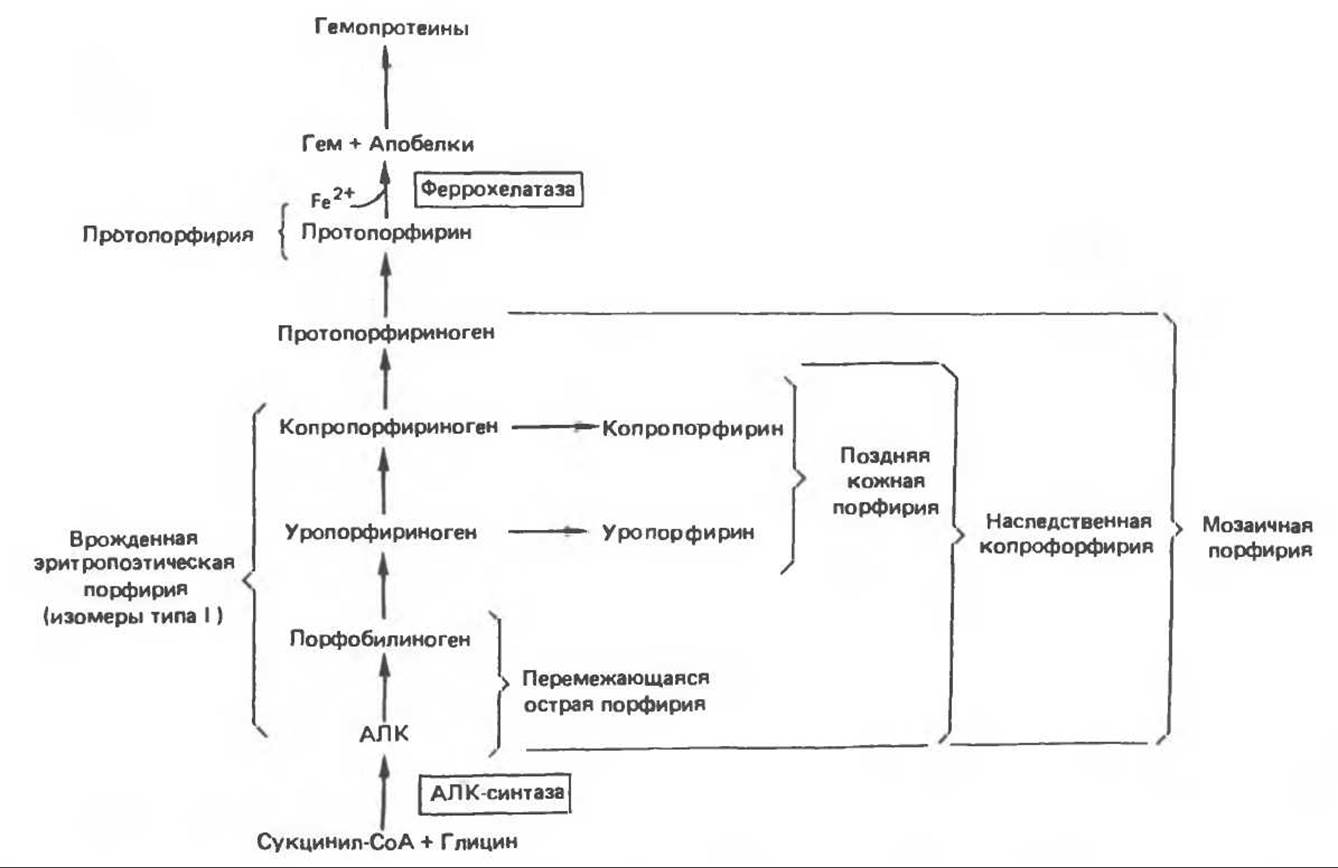
Рис. 33.11. Последовательные стадии биосинтеза гема, с указанием предшественников, экскретируемых с мочой, при различных формах порфирин Фигурные скобки объединяют соединения, которые экскретируются с мочой в избыточных количествах при обострении указанных форм порфирии. АЛК — 5-аминолевулиновая кислота. (Воспроизведено с изменениями из статьи Kaufman L., Merver H.S. Biochemical defects in two types of human hepatic prphyria. N. Engl. J. Med 1970:283:954.)
Врожденная эритропоэтическая порфирия — это еще более редкое врожденное заболевание, наследуемое по аутосомно-рецессивному типу. Молекулярная природа этой болезни точно неизвестна; установлено, однако, что для нее характерен определенный дисбаланс относительных активностей уропорфириноген-ІІІ-косинтазы и уропорфириноген-І-синтазы. Образование уропорфириногена I в количественном отношении значительно превосходит синтез уропорфириногена III — нормального изомера на пути синтеза гема. Хотя генетическое нарушение распространяется на все клетки, проявляется оно по неизвестной причине преимущественно в эритропоэтической ткани. Пациенты с врожденной эритропоэтической порфирией экскретируют большие количества изомеров типа I уропорфириногена и копропорфириногена; в моче оба этих соединения самопроизвольно окисляются в уропорфирин 1 и копропорфирин I — красные флуоресцирующие пигменты. Сообщалось о случае, когда наблюдалось небольшое повышение концентрации уропорфирина III, но отношение изомеров типа I и III составляло примерно 100:1. Циркулирующие эритроциты содержат большое количество уропорфирина I, однако наивысшая концентрация этого порфирина отмечена в клетках костного мозга (но не в гепатоцитах).
Вероятно, вследствие образования меньших количеств истинного предшественника гема, уропорфириногена III, и возникающего вследствие этого относительного дефицита гема в эритропоэтических тканях больных происходит индукция АЛК-синтазы. Эта индукция приводит к перепроизводству порфириногенов типа I. Наряду с усилением синтеза АЛК-синтазы и перепроизводством порфириногенов типа I повышаются образование и экскреция порфобилиногена и АЛК. Таким образом, на основе биохимических отклонений можно предсказывать появление клинических симптомов, сходных с теми, которые наблюдаются при ПОП, но помимо этого отмечается светочувствительность кожи, обусловленная характером спектра поглощения порфириновых соединений, которые образуются в больших количествах. У пациентов отмечаются трещины на коже, часто наблюдаются гемолитические явления.
Наследственная копропорфирия — аутосомно-доминантное нарушение, обусловленное дефицитом копропорфириноген-оксидазы — митохондриального фермента, ответственного за превращение копропорфириногена III в протопорфириноген IX. Копропорфириноген III в больших количествах удаляется из организма в составе фекалий, а также вследствие его растворимости в воде экскретируется в большом количестве с мочой. Как и уропорфириноген, копропорфириноген на свету и воздухе быстро окисляется, превращаясь в красный пигмент копропорфирин.
Ограниченная при этом заболевании способность к синтезу гема (особенно в стрессовых условиях) приводит к дерепрессии АЛК-синтазы. В результате наблюдается избыточное образование АЛК и порфобилиногена, а также других интермедиатов на пути синтеза гема, образующихся на стадиях, предшествующих наследственно заблокированному этапу. Соответственно у пациентов с наследственной копропорфирией обнаруживаются все признаки и симптомы, связанные с избытком АЛК и порфобилиногена, которые характерны для перемежающейся острой порфирии, но помимо этого у них имеется повышенная светочувствительность, обусловленная присутствием избыточных количеств копропорфириногенов и уропорфириногенов. При этом заболевании введение гематина также может вызвать по крайней мере частичную репрессию АЛК-синтазы и смягчение симптомов, обусловленных перепроизводством интермедиатов биосинтеза гема.
Мозаичная порфирия, или наследственная фотокопропорфирия, является аутосомно-доминантным нарушением, при котором происходит частичное блокирование ферментативного превращения протопорфириногена в гем. В норме это превращение осуществляется двумя ферментами, протопорфириногеноксидазой и феррохелатазой, локализованными в митохондриях. Судя по данным, полученным на культуре фибробластов кожи, у больных мозаичной порфирией содержание протопорфириногеноксидазы составляет лишь половину нормального количества. У пациентов с мозаичной порфирией наблюдается относительная недостаточность содержания гема в стрессовых условиях, а также дерепрессированное состояние печеночной АЛК-синтазы. Как отмечалось выше, повышенная активность АЛК-синтазы ведет к перепроизводству всех интермедиатов синтеза гема на участках перед заблокированной стадией. Таким образом, пациенты с мозаичной порфирией экскретируют с мочой избыточные количества АЛК, порфобилиногена, уропорфирина и копропорфирина, а с фекалиями выделяют уропорфирин, копропорфирин и протопорфирин. Моча больных пигментирована и флуоресцирует, а кожа чувствительна к свету так же, как и у больных поздней кожной порфирией (см. ниже).
Поздняя кожная порфирия, вероятно, является наиболее распространенной формой порфирии. Обычно она связана с теми или иными поражениями печени, особенно при избыточном потреблении алкоголя или перегрузке ионами железа. Природа метаболического нарушения точно не установлена, но вероятной причиной является частичная недостаточность уропорфириноген-декарбоксилазы. Нарушение, по-видимому, передается как аутосомно-доминантный признак, но генетическая пенетрантность различна и в большинстве случаев зависит от наличия нарушений функций печени. В соответствии с предсказаниями моча содержит повышенные количества уропорфиринов типа I и III; в то же время экскреция с мочой АЛК и порфобилиногена наблюдается сравнительно редко. Иногда моча содержит весьма значительное количество порфиринов, придающих ей розоватый оттенок; при подкислении она чаще всего дает в ультрафиолетовой области розовую флуоресценцию.
Печень содержит большие количества порфиринов и поэтому сильно флуоресцирует, тогда как у эритроцитов и клеток костного мозга флуоресценция отсутствует. Главным клиническим проявлением при поздней кожной порфирии является повышенная светочувствительность кожи. У больных не наблюдается ни повышенной активности АЛК-синтазы, ни соответственно избыточного содержания в моче порфобилиногена и АЛК; это коррелирует с отсутствием острых приступов, характерных для перемежающейся острой порфирии.
Протопорфирин, или эритропоэтическая протопорфирия, по-видимому, обусловлена доминантно наследуемой недостаточной активностью феррохелатазы в митохондриях всех тканей; клинически эта болезнь проявляется как острая крапивница, вызываемая воздействием солнечных лучей. Эритроциты, плазма и фекалии содержат повышенные количества протопорфирина IX, а ретикулоциты (незрелые эритроциты) и кожа (при исследовании с помощью биопсии) часто флуоресцируют красным светом.
Печень, вероятно, тоже вносит вклад в повышение образования протопорфирина IX, однако экскреции с мочой порфиринов и их предшественников не наблюдается.
Приобретенная (токсическая) порфирия может быть вызвана действием токсических соединений, таких, как гексахлорбензол, соли свинца и других тяжелых металлов, а также лекарственными препаратами, например гризеофульвином. Тяжелые металлы являются ингибиторами ряда ферментов в системе синтеза гема, включая АЛК-дегидратазу, уропорфириноген-синтазу и феррохелатазу.