Биохимия - Химические реакции в живой клетке Том 2 - Д. Мецлер 1980
Типы реакций, катализируемых ферментами
Реакции замещения у карбонильных групп
Химотрипсин и трипсин
Наиболее подробно изученная протеиназа, химотрипсин, существует в нескольких слегка различающихся формах, которые образуются в результате расщепления определенных пептидных связей в молекуле химотрипсиногена. Последняя представляет собой единую полипептидную цепь, построенную из 245 аминокислот; аминокислоты в активном ферменте обычно нумеруются в соответствии с их положением в исходном зимогене. Важную роль в выяснении механизма действия химотрипсина сыграли данные, полученные при изучении ацетилхолинэстеразы. Было показано, что этот ключевой фермент нервной системы необратимо инактивируется группой сильных фосфорсодержащих ядов, используемых как инсектициды и как отравляющие газы нервно-паралитического действия.
а. Серин в активном центре
В 1949 г. было показано, что один из ядов нервно-паралитического действия, диизопропилфторфосфат (ДФФ), инактирует химотрипсин.
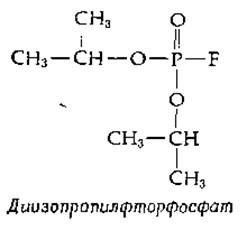
Если на химотрипсин воздействовали диизопропилфторфосфатом, меченным 32Р, то 32Р оказывался прочно связанным с ферментом ковалентной связью. Дальнейшие эксперименты показали, что при денатурации меченого фермента и последующем его гидролизе кислотой высвобождения 32Р не происходит. Образующийся радиоактивный фрагмент был идентифицирован как О-фосфосерин. С химической точки зрения реакция между диизопропилфторфосфатом и химотрипсином протекает просто и состоит в следующем.
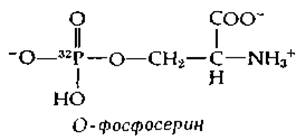
Гидроксильная группа боковой цепи серинового остатка атакует молекулу диизопропилфторфосфата у атома фосфора, замещая атом фтора. Заметим, что эта реакция представляет собой реакцию нуклеофильного замещения у атома фосфора (реакция типа 1.В, табл. 7-1), но в этом случае она протекает на ферменте, обычно катализирующем реакцию замещения у атома углерода карбонильной группы. Молекулу диизопропилфторфосфата можно рассматривать как псевдосубстрат (квазисубстрат), который реагирует с ферментом аналогично настоящему субстрату, но который не обеспечивает нормального завершения реакции.
На основании изучения пептидов, образующихся при частичном гидролизе меченного 32Р химотрипсина, была определена последовательность аминокислот, окружающих реакционноспособный остаток серина, и в конечном счете было установлено, что в общей последовательности цепи этот остаток серина занимает положение 195. Предшествующие положения (193 и 194) занимают глицин и аспарагиновая кислота, а в положении 196 находится еще одна молекула глицина
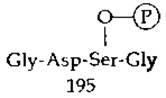
Такая же последовательность аминокислот в окружении реакционноспособных остатков серина была вскоре обнаружена в трипсине, тромбине, эластазе и трипсиноподобном ферменте коконазе, используемом тутовым шелкопрядом для освобождения из кокона [27]. Близкая по химической структуре последовательность, содержащая Glu-Ser-Ala, была обнаружена в ацетилхолинэстеразе. Таким образом, существует семейство сериновых пептидаз и эстераз, для которых характерна общая последовательность аминокислот вокруг реакционноспособного серина и способность ингибироваться диизопропилфторфосфатом [28].
б. Промежуточное соединение ацилфермент. Другой псевдосубстрат, n-нитрофенилацетат,
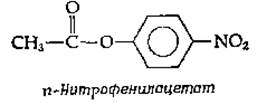
реагирует с химотрипсином при pH 4 (гораздо ниже pH-оптимума скорости реакции гидролиза) с быстрым выделением n-нитрофенола и образованием ацетилпроизводного фермента. Образующийся ацетилфермент очень медленно гидролизуется при pH 4, но гораздо быстрее при более высоких значениях pH. Эти эксперименты позволяют предположить, что химотрипсин, подобно сахарозофосфорилазе, действует по механизму двойного замещения:
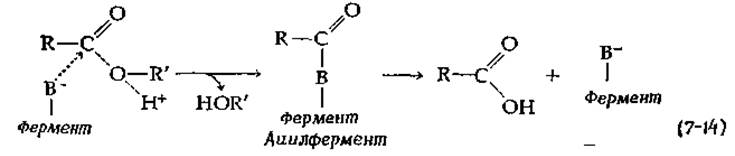
Что представляет собой группа —В- в этом уравнении? Опыты с диизопропилфторфосфатом указывают на то, что —O- -группа принадлежит Ser-195. Однако этому предположению противоречит очень слабая кислотность —СН2ОН-группы. Кроме того, согласно другим данным, роль группы —В- выполняет имидазольная группа гистидина. Например, известно, что каталитическая активность химотрипсина при гидролизе сложных эфиров меняется с изменением pH. В то время как константа Михаэлиса мало меняется с изменением pH, зависимость Vmах от pH графически представлена типичной колоколообразной кривой (рис. 6-13) с оптимумом между pH 8 и 9. Из характера кривых для ряда различных эстераз найдены два значения рKа: одно из них находится между 6,1 и 6,8, а второе — между 9,0 и 9,6. Значение рKа, лежащее между 6,1 и 6,8, логичнее всего было бы приписать имидазольной группе гистидина, которая в каталитически активной форме фермента находится в непротонированном состоянии. Имеются и другие эксперименты, указывающие на участие в каталитическом процессе остатка гистидина. Например, химотрипсин реагирует с 2,4-динитрофторбензолом, и атака этим реагентом одного из двух остатков гистидина в молекуле фермента приводит к его инактивации.
в. Неферментативные модели
Было показано, что соединения, содержащие имидазол, катализируют неферментативный гидролиз n-нитрофенилацетата с образованием в качестве промежуточных продуктов нестойких ацетилимидазолов:
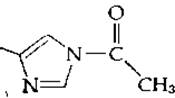
Было очень соблазнительно допустить, что ацилферментные промежуточные соединения, представленные в уравнении (7-14), могут иметь такое же строение. Таким образом, имелось два кандидата на роль группы —В: серин и гистидин. Хотя не вызывало сомнения то, что стабильные конечные продукты реакции с псевдосубстратами — это, несомненно, производные серина, все же оставалась возможность, что они являются побочными продуктами и что гистидин участвует в образовании промежуточных соединений, быстро образующихся и быстро распадающихся.
г. Трехмерная структура
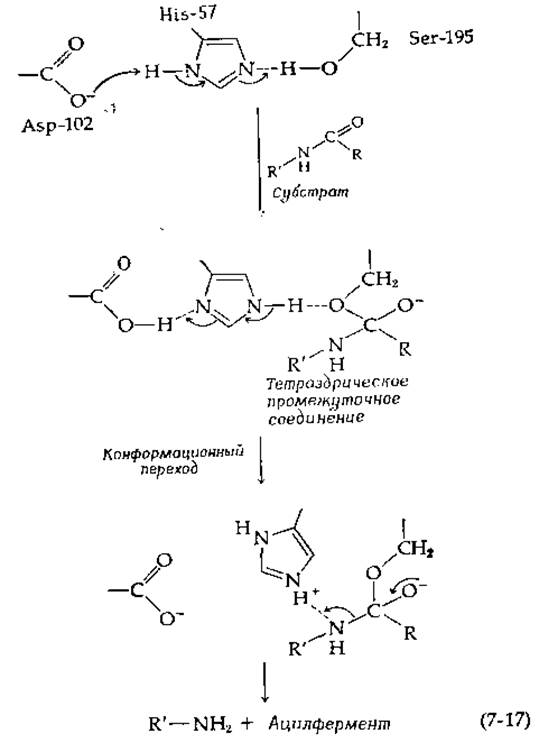
В настоящее время структура химотрипсина и трипсина расшифрована благодаря использованию метода дифракции рентгеновских лучей [29—32], подтвердившего предположения, сделанные на основании химических исследований. Как Ser-195, так и His-57 находятся в активном центре ферментов (рис. 7-2). Следует иметь в виду, что метод Дифракции рентгеновских лучей кристаллом фермента не дает возможности обнаружить положение атомов водорода в молекуле фермента и что на рисунке они проставлены согласно химической логике. Так, короткое расстояние (0,30 нм) между азотом остатка His-57 и кислородом остатка Ser-195 свидетельствует о наличии водородной связи. Аналогичные рассуждения привели к выводу о присутствии других водородных связей, показанных на рисунке. Если гистидин находится в непротонированной форме, а гидроксильная группа серина протонирована, то мы видим, что гистидин может выступать в роли акцептора протона от —СН2ОН-группы серина (т. е. в роли общего основного катализатора), повышая нуклеофильность кислорода гидроксильной группы.
Исследования по дифракции рентгеновских лучей показали, что His-57 образует также водородную связь с карбоксильной группой остатка Asp-102, которая в свою очередь образует водородные связи с двумя другими группами. Карбоксильная группа боковой цепи остатка Asp-102 является одной из немногих карбоксильных групп, замаскированных в глубине белковой молекулы. Блоу [29] считает, что в такой структуре должна иметься система переноса заряда (a charge-relay system), с помощью которой протоны могут синхронно перемещаться от Ser-195 к имидазолу и от имидазола к Asp-102. Значение этой системы для сериновых протеиназ полностью еще не установлено, но этот вопрос следует рассматривать в свете концепции «таутомерного катализа» (гл. 6, разд. Д,5, е). Возможно также, что перенос заряда обусловлен смещениями электронной плотности в пептидной цепи. То обстоятельство, что такая же система переноса зарядов в ходе эволюции возникла в другой, в остальном совершенно отличной по действию сериновой протеиназе, субтилизине (из Bacillus subtilis), позволяет сделать заключение, что эта система может действительно иметь существенное значение для механизма ферментативного катализа [33, 34].
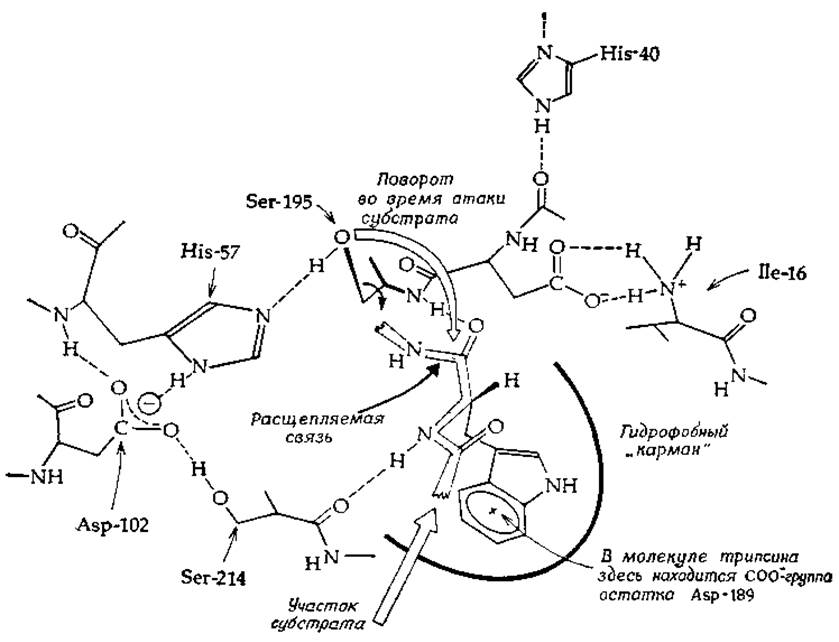
РИС. 7-2. Активный центр химотрипсина с фрагментом связанного субстрата (по Блоу [29] и Хендерсону и Янгу [13]).
Прямые наблюдения методом ЯМР протона, участвующего в образовании водородной связи между His-57 и Asp-102, подтверждают концепцию о переносе заряда [35]. Однако попытки сконструировать эффективную неферментативную модель, включающую систему переноса заряда, оказались безуспешными [36].
Повышение нуклеофильности гидроксильной группы серина — не единственная возможная роль остатка His-57. Группа R'—NH-, вытесняемая из субстрата, является плохой уходящей группой, если ее не перевести в протонированную форму (общий кислотный катализ):
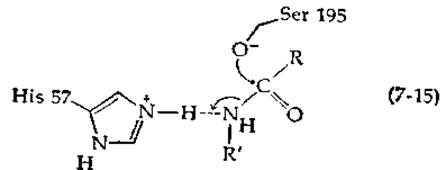
Как указывал Дженкс [37], часто бывает трудно провести различие между общим основным и общим кислотным катализом. Возможно, His-57 выполняет сразу обе функции, сперва оттягивая протон от гидроксильной группы серина, а затем отдавая протон уходящей группе [13, 38].
Другой вероятный механизм участия фермента в реакции замещения у карбонильной группы состоит в протонировании карбонильного кислорода кислотной группой фермента:
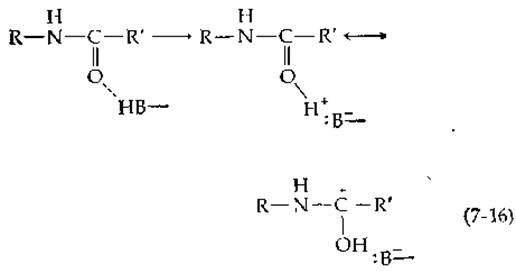
Это должно привести к значительному увеличению положительного заряда атома углерода, что в свою очередь должно облегчить нуклеофильную атаку. Такое взаимодействие должно также стабилизировать тетраэдрическое промежуточное соединение [уравнение (7-13)]. Карбонильный кислород проявляет очень слабую основность, но он может протонироваться подходящим образом ориентированной кислотной группой фермента [НВ в уравнении 7-16)]. В сериновых протеиназах эта функция, очевидно, выполняется двумя NH-группами амидных связей, одна из которых в химотрипсине принадлежит остатку Ser-195 (рис. 7-2). По-видимому, подгонка субстрата к полости оксианиона между двумя NH-группами хорошо выполняется только для тетраэдрического промежуточного соединения [33].
Было высказано предположение [38], что образование тетраэдрического оксианиона (с зарядом, передаваемым от замаскированного карбоксилата остатка Asp-102 к субстрату) инициирует конформационный переход, благодаря которому при распаде тетраэдрического промежуточного соединения имидазольная группа остатка His-57 получает возможность принять обратно протон от Asp-102 и протонировать уходящую группу [уравнение (7-17)].
Почему происходит конформационное изменение? В молекуле фермента имеется сложная сеть водородных связей. Появление отрицательного заряда на атоме кислорода тетраэдрического промежуточного соединения должно обязательно повлиять на распределение электронных плотностей внутри некоторых водородных связей, отдаленных от активного Центра. Если молекула белка может существовать в нескольких энергетически эквивалентных конформациях, то возможна ситуация, когда изменение в распределении зарядов вызывает резкий переход из одного конформационного состояния в другое, причем подобный конформационный переход может являться важной составной частью каталитического процесса.
При высоких значениях pH скорость действия химотрипсина падает, и характер pH-зависимости указывает на существование в активном центре группы С рKа от ~ 8 до 9. Это значение рКа может относиться к N-концевой аминогруппе Ilе-16. Аминогруппа Ilе-16 участвует в образовании одной из связей, расщепляемых при превращении зимогена в активный фермент. Эта аминогруппа образует ионную связь (ионную пару) с остатком Asp-194 (рис. 7-2), который находится рядом с серином активного центра. Возможно, ионная связь способствует поддержанию фермента в нужной для реакции конформации. Депротонирование при pH выше 8—9 должно вызывать инактивацию [39].
Другая возможность состоит в том, что группа, имеющая высокое значение рKа, принадлежит второму гистидину фермента — His-40. Однако этот гистидин отсутствует в бактериальной сериновой протеиназе, имеющей тот же самый тип pH-зависимости. His-40 расположен вблизи от остатка Asp-194 и связан водородной связью с пептидным карбонилом. Считают, что он может играть определенную роль во взаимопревращениях зимоген — активный фермент.
д. Субстратная специфичность
Подобно большинству ферментов, трипсин и химотрипсин проявляют четко выраженную специфичность по отношению к определенным субстратам. Быстрое расщепление химотрипсином наблюдается в том случае, когда С=О-группа расщепляемой пептидной связи принадлежит одной из ароматических аминокислот. Таким образом, центр фермента, связывающий субстрат, должен иметь участок, осуществляющий связывание преимущественно больших, плоских ароматических групп. Анализ кристаллической структуры фермента показывает, что этот центр (рис. 7-2) состоит из гидрофобных боковых цепей аминокислот. В трипсине же участок активного центра, отвечающий за специфическое связывание субстратов, содержит фиксированный отрицательный заряд, обусловленный карбоксильной группой боковой цепи Asp-189. Этим объясняется тот факт, что трипсин расщепляет только те пептидные связи, к которым примыкают остатки аргинина или лизина, несущие в нейтральной среде положительный заряд [32].