Биохимия - Химические реакции в живой клетке Том 3 - Д. Мецлер 1980
Свет в биологии
Поглощение света веществом
Инфракрасные спектры
Поглощение в ближней инфракрасной области определяется переходом молекулы с одного колебательного уровня на другой. Типичной частотой является частота, соответствующая максимуму полосы поглощения «амид А» — 3300 см-1 (длина волны 3,0 мкм), что отвечает примерно 1014 с-1. Анализ инфракрасных спектров обычно начинается с рассмотрения валентных колебаний двухатомной молекулы. Представим, что два ядра молекулы соединены пружинкой. Колебательную энергию такой молекулы можно рассматривать как энергию гармонического осциллятора. Согласно квантовомеханическому подходу, энергия осциллятора принимает только дискретные значения, а соответствующие энергетические уровни располагаются на одинаковом расстоянии друг от друга, равном hv, где v — частота кванта света, поглощение которого повышает энергию до значения, соответствующего следующему энергетическому уровню. В основном (невозбужденном) состоянии молекула уже обладает «энергией нулевых колебаний», равной половине энергии, необходимой для перехода на следующий уровень.
Гармонический осциллятор служит удобной моделью для описания поведения молекул, находящихся только на нижних колебательных уровнях; в состояниях с более высокой энергией наблюдаются существенные отклонения от модели. На нижних энергетических уровнях изменение расстояния между центрами атомов в ходе колебаний составляет ±10%, с повышением же энергии - это расстояние увеличивается и движение становится ангармоническим. Энергетические состояния молекул часто изображают в виде кривых Морза, представляющих собой зависимость энергии от расстояния между ядрами (рис. 13-2). Когда это расстояние становится слишком малым, энергия резко возрастает. По мере растяжения связи наступает момент, когда дальнейшее небольшое увеличение энергии приводит к разрыву связи и к распаду двухатомной молекулы на атомы (или более сложной молекулы на фрагменты). Колебательные энергетические уровни изображают в виде горизонтальных линий, проведенных на соответствующих высотах на графике Морза (рис. 13-2)
Поскольку каждому колебательному энергетическому уровню соответствует множество вращательных подуровней, инфракрасные спектры представляют собой набор полос поглощения, что обусловлено одновременным изменением колебательной и вращательной энергии молекул. Таким образом, вместо отдельных линий, отвечающих переходам между колебательными уровнями, наблюдаются серии резких близко расположенных линий. Примером такого рода служит полоса поглощения, соответствующая валентному колебанию связи Н—Сl в газообразном НСl с максимумом 2886 см-1 (3,46 мкм): на самом деле она представляет собой набор почти равноотстоящих линий, расположенных по обе стороны от указанной основной частоты в интервале от ~2600 до — 3100 см-1. Расстояние между соседними линиями равно ~21 см-1, что соответствует вращательной частоте, находящейся в микроволновой области шкалы электромагнитных волн (Герцберг [8], стр. 55). При снятии спектра с низким разрешением наблюдается одна широкая полоса1).
Расшифровка инфракрасных спектров двухатомных молекул не составляет особого труда, но полосы поглощения более сложных соединений уже не удается отнести к колебаниям определенных химических связей. В этом случае говорят об основных (нормальных) колебаниях молекулы. Основными называют такие колебания, при которых положение центра тяжести молекулы не меняется. Для молекулы из п атомов число таких колебаний равно 3n—6. Хотя при этом нередко преобладает колебание какой-то одной связи, возможно синхронное движение многих атомов. При описании основных колебаний молекулы пользуются такими терминами, как растяжение, изгиб (в плоскости или с выходом из плоскости), кручение и деформация. Все 3n—6 полос в инфракрасном спектре наблюдаются довольно редко. Объясняется это отчасти тем, что некоторые колебания не сопровождаются изменением дипольного момента (например, симметричное растяжение линейной молекулы СO2). Другие же полосы оказываются попросту слишком слабыми, чтобы их можно было четко зарегистрировать.
Частота колебаний, захватывающих сразу много атомов молекулы, — так называемых скелетных колебаний — как правило, лежит в области 700—1400 см-1 (14—7 мкм). Частоты же колебаний, определяемых главным образом конкретными функциональными группами, составляют обычно 1000—5000 см-1 (10—2 мкм). Так, например, валентные колебания С—Н-, N—Н- и О—Н-связей имеют характерные частоты, равные, как правило, ~2900, 3300 и 3600 см-1 соответственно. Отметим ряд важных фактов. Энергия (и частота) колебаний возрастает с увеличением различия в электроотридательности двух атомов, образующих связь. Чем больше масса атомов, тем ниже частота (например, частота колебаний С—О в первичном спирте составляет ~1053 см-1). Частота колебаний двойной связи выше, чем одинарной (так, для связи С = O она равна — 1700 см-1; эти валентные колебания соответствуют одной из самых интенсивных полос поглощения инфракрасного спектра). Сильный и весьма характерный эффект оказывает образование водородных связей: частота колебаний связи О—Н, составляющая ~3600 см-1, при образовании водородной связи уменьшается до 3500 см-1.
1) Это лишь одна из причин уширения полос поглощения в инфракрасной области в растворе; другая обусловлена взаимодействием с растворителем, в результате которого поглощающие молекулы находятся в неэквивалентном окружении.
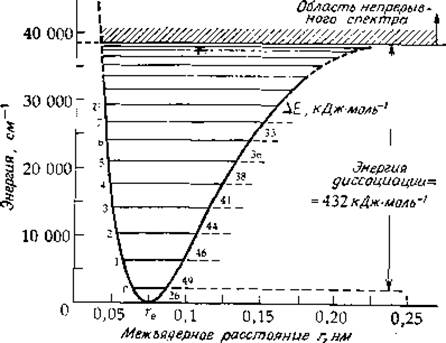
РИС. 13-2. Зависимость потенциальной энергии молекулы водорода от расстояния между ядрами; указаны колебательные энергетические уровни молекулы. ∆Е — разность энергий соседних энергетических уровней; а — колебательные квантовые числа ([5], стр. 135.)
Согласно теории гармонического осциллятора, разрешенными являются переходы с данного колебательного энергетического уровня лишь на соседний более высокий уровень, однако для ангармонических осцилляторов возможны слабые переходы на более высокие колебательные уровни. В результате возникают «обертоны», частота которых приблизительно кратна основной частоте. Кроме того, могут наблюдаться полосы, частоты которых равны сумме или разности частот отдельных инфракрасных полос. Эти полосы очень слабые, но тот факт, что они лежат в области относительно высоких энергий в ближней инфракрасной области (4000—12 500 см-1), позволяет выявить их даже легче, чем основные полосы, располагающиеся очень близко друг к другу в инфракрасной области.
а. Колебательные частоты амидных групп
Поскольку амидные группы присутствуют как в белках, так и в пуриновых и пиримидиновых основаниях, их инфракрасные полосы поглощения привлекли к себе большое внимание. Из множества полос поглощения указанной группы (описанных в книге Фрэзера и Мак-Рэе [11]) особый интерес представляют три полосы.
Полоса амид 1 при ~ 1680 см-1 соответствует нормальным колебаниям в плоскости амидной группы и прежде всего валентным колебаниям связи С = О. Полоса амид II при 1500 см-1 тоже обусловлена колебаниями в пределах плоскости, включая изгиб N—Н-связи, тогда как полоса амид А с более высокой частотой, равной ~3450 см-1, обусловлена валентными колебаниями N—Н-связи. Участие N—Н-группы в образовании водородной связи сдвигает полосу амид А к ~3300 см-1. Инфракрасный спектр белка [12], содержащий полосы амид А, амид I и амид II, приведен на рис. 13-3. Обратите внимание на сложную форму полос. Форма полосы амид I сильно зависит от конформации пептидной цепи. Согласно эмпирическому правилу, частота полосы поглощения амидных групп в а-спиралях на 20 см-1 больше, чем частота соответствующих полос в ß-структурах. Правда, более тщательный анализ нормальных колебаний пептидных цепей привел к иным выводам [11, 13—15].
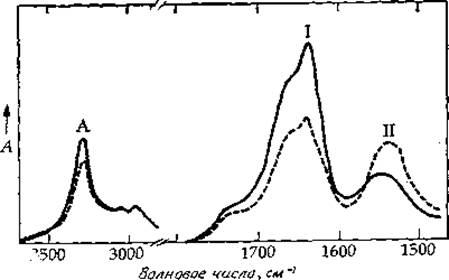
РИС. 13-3. Инфракрасный дихроизм фибрилл инсулина. Сплошная линия: вектор напряженности электрического поля параллелен оси фибриллы; штриховая линия: вектор напряженности электрического поля перпендикулярен оси фибриллы. [Burke М. J., Rougvie М. A., Biochemistry, 11, 2437 (1972)].
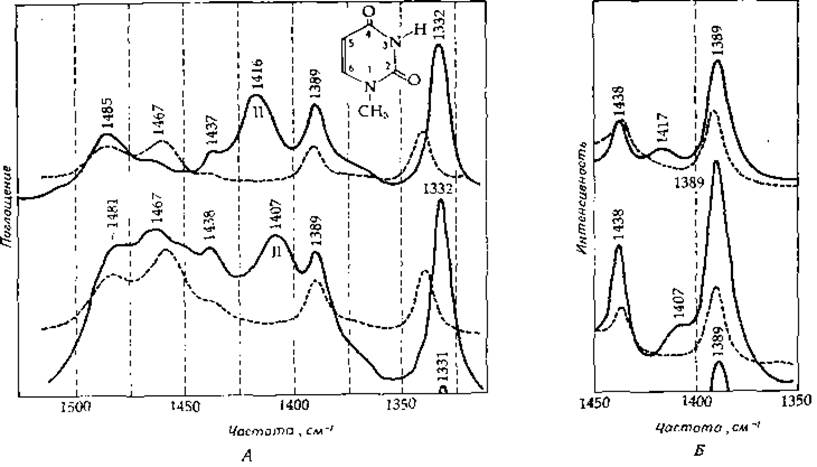
РИС. 13-4. Инфракрасный (А) и рамановский (Б) спектры 1-метилурапила в Н2О (сплошная линия) и D2O (штриховая линия). Приведены спектры обычного 1-метил-урацила (вверху) и его специфически меченного производного, содержащего 18O в положении 4 (внизу) [16].
Одним из наиболее распространенных методов исследования ориентированных пептидных цепей является метод инфракрасного дихроизма. При этом регистрируют спектры поглощения белка для двух взаимно перпендикулярных направлений поляризации падающего света. В одном случае вектор напряженности электрического поля параллелен пептидным цепям, а в другом — перпендикулярен им. Такая пара спектров для ориентированных фибрилл инсулина приведена на рис. 13-3. Считается, что молекулы инсулина находятся в этом случае в ß-конформации и уложены поперек оси фибриллы (Kpocc-ß-структура). Таким образом, когда вектор напряженности электрического поля параллелен оси фибриллы, он перпендикулярен пептидным цепям. Поскольку полоса амид I определяется прежде всего колебаниями карбонильной группы, которые в ß-структуре перпендикулярны пептидным цепям, интенсивность этой полосы больше для случая, когда вектор напряженности электрического поля тоже перпендикулярен пептидным цепям, чем для случая, когда этот вектор им параллелен (перпендикулярен оси фибриллы; рис. 13-3). То же самое справедливо и для полосы амид А, которая определяется в основном растяжением связи N—Н. Дихроизм полосы амид II носит противоположный характер, поскольку здесь определяющую роль играет изгиб N—Н-связи, который осуществляется в пределах плоскости пептидной группы, но происходит в продольном направлении.
Инфракрасная спектроскопия нашла применение и при анализе полос поглощения амидных групп пиримидинов [16]. На рис. 13-4, А приведен спектр 1-метилурацила в Н2О и D2О. Заметим, что в D2О полоса амид II вообще отсутствует. Это иллюстрирует еще один путь применения инфракрасной спектроскопии, который оказался особенно полезен при изучении белков. Исчезновение полосы амид II при перенесении белка в D2О дает возможность проследить за обменом протонов, участвующих в образовании водородных связей, в структурированных областях белков [10]. На рис. 13-4 приведен также инфракрасный спектр 1-метилурацила, содержащего 180 в 4-м положении. Обратите внимание на сдвиг полосы амид II на 7 см-1, указывающий, что колебания, связанные с изгибом N—Н-связи, в значительной мере сопряжены с валентными колебаниями связей С = О и С = С.
б. Раман-спектры
Рамановская спектроскопия основана на исследовании спектров рассеяния света. При столкновении фотона с молекулой может иметь место упругое соударение, при котором фотон не теряет энергию, но изменяет направление своего движения. Такое рассеяние известно под названием рэлеевского и лежит в основе метода определения молекулярных весов соединений. Соударения могут быть также неупругими; они характеризуются тем, что энергия молекулы и фотона изменяется. Поскольку эти изменения носят квантовый характер и определяются колебательными и вращательными уровнями молекулы, анализ спектра рассеянного света (спектра Рамана) дает почти ту же информацию, что и обычный инфракрасный спектр. Необходимо, однако, помнить один момент: правила отбора в этих двух случаях различаются. В инфракрасной спектроскопии разрешены одни переходы, в раман-спектроскопии — другие. Таким образом, имеет смысл снять и тот и другой спектр исследуемого образца. До недавнего времени раман-спектроскопия находила весьма ограниченное применение из-за малой интенсивности рассеянного света. Однако использование для возбуждения лазеров существенно повысило ценность указанного метода [16—20]. В качестве примера на рис. 13-4, Б приведен раман-спектр 1-метилурацила. Заметим, что интенсивность полосы амид II (относительно полосы амид I) в раман-спектре значительно меньше, чем в инфракрасном спектре поглощения. Особый интерес представляет резонансная раман-спектроскопия [19—21], где используется лазерный пучок с длиной волны, соответствующей длине волны электронного перехода. Рассеяние света при этом часто существенно усиливается на частотах, которые отличаются от частоты лазера на частоту рамановского рассеяния, происходящего на группах хромофора или на группах молекулы, соседствующей с хромофором. Несмотря на определенные экспериментальные трудности, указанный метод позволяет изучать структурные особенности какого-либо конкретного участка макромолекулы.