Пептидная саморегуляция живых систем (факты и гипотезы) - Шатаева Л. К. 2003
Пептиды в водных растворах
Особенности строения аминокислот и олигопептидов
Как известно, белки состоят из а-аминокарбоновых кислот, обычно называемых аминокислотами. Их структуры представлены в любом учебнике биохимии или справочнике. В растительном и животном мире к настоящему времени обнаружено более 100 аминокислот различного строения, но только 20 из них кодируются генетическим кодом, т. е. для каждой из этих 20 аминокислот имеется определенное сочетание нуклеотидных звеньев (кодонов), которое определяет место аминокислоты в полипептидной цепи при рибосомальном синтезе. В настоящей монографии мы рассмотрим только кодируемые аминокислоты, которые составляют основу биологических процессов на Земле. Их строение имеет определенные пространственные и электрохимические особенности.
1. Все аминокислоты, кроме глицина, обладают хиральностью, т. е. имеют две зеркально симметричные пространственно несовместимые формы L и D (рис. 1, А). Аминокислоты, входящие в состав природных пептидов и белков, имеют, как правило, L-форму; исключения встречаются в пептидных антибиотиках животного происхождения (Kreil, 1997).
2. Аминогруппа имеет явно выраженный основный характер, а карбоксильная группа — кислый: для глицина значения константы кислотной диссоциации (рКа) равны 9.6 и 2.3 соответственно. В водном растворе в физиологических условиях протон карбоксильной группы переходит к аминогруппе (рис. 1, Б) и молекула превращается в цвиттерион (Полинг, 1964). Благодаря фиксированному в пространстве расположению положительного и отрицательного зарядов аминокислоты в воде имеют постоянный дипольный момент.
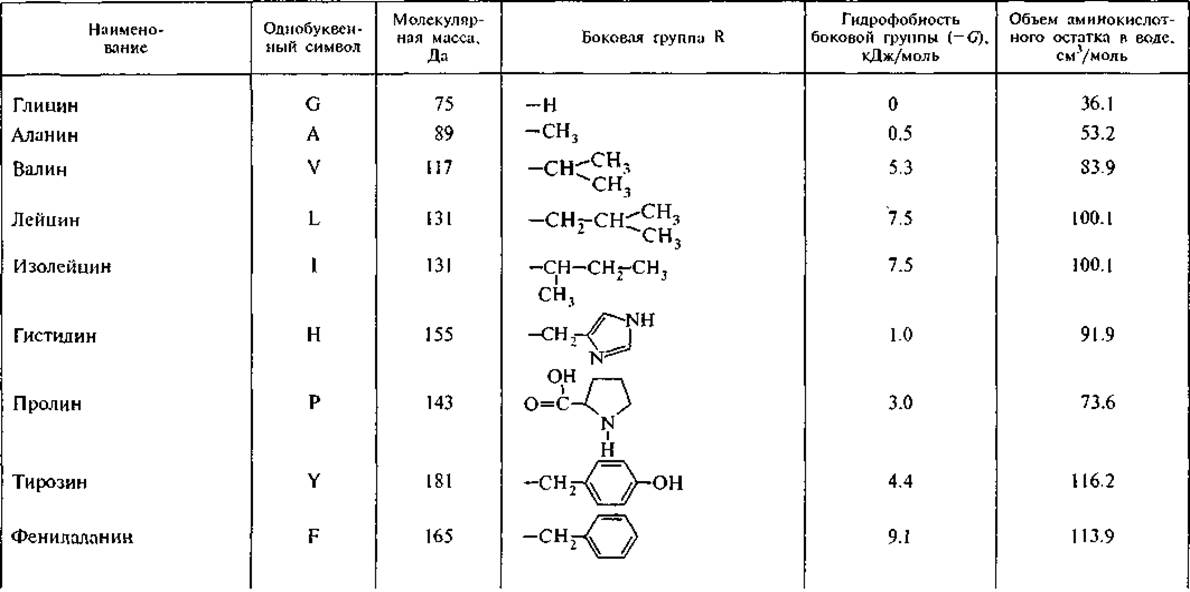
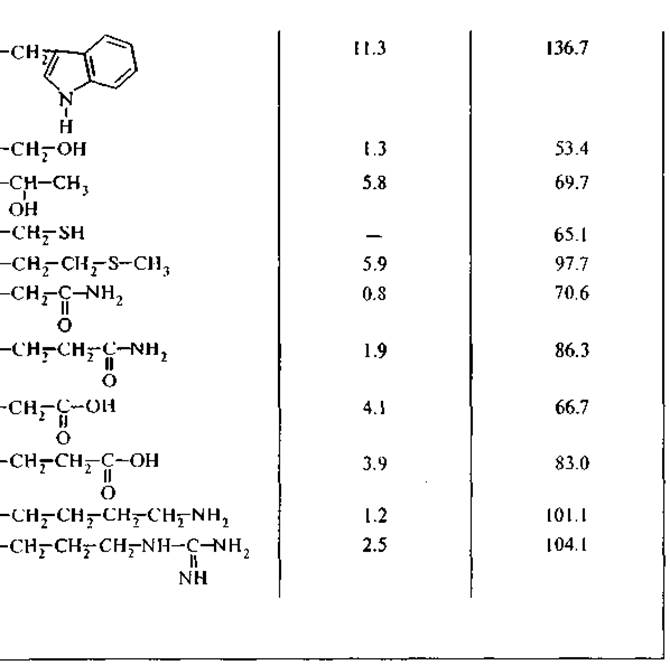
3. R — боковые группы аминокислот (рис. 1, А) имеют широкий диапазон гидрофильных и гидрофобных свойств (табл. 1), которые определяют межмолекулярные взаимодействия аминокислот. Боковые группы лизина и аргинина проявляют свойства сильных оснований (значения рКа равны 10.5 и 12.5 соответственно); боковые группы аспарагиновой и глутаминовой кислот проявляют выраженные кислотные свойства (рКа равно 3.6 и 4.2 соответственно).
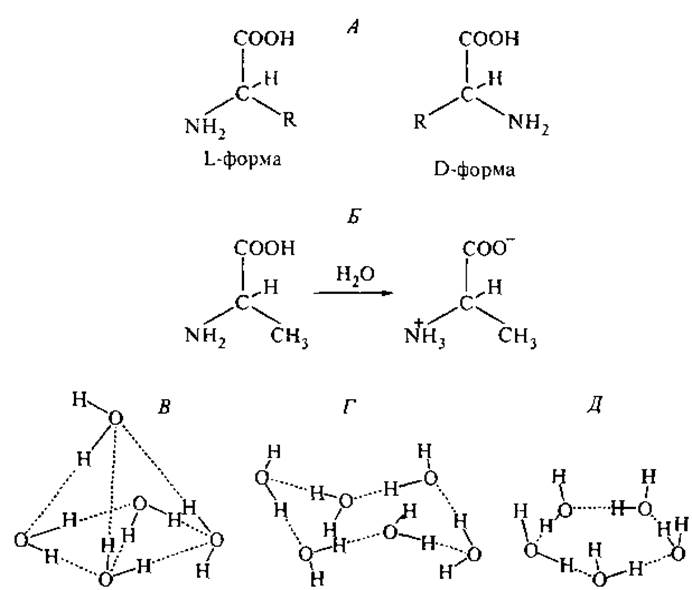
Рис. 1. Пространственные структуры аминокислот и водного окружения. А — стереоизомеры а-карбоновых аминокислот, где R — боковая группа; Б — цвит-терионная структура L-аланина в водном растворе при pH 7.0. В—Д — координация молекул воды с помощью водородных связей, определяющая гидратацию гидрофильных и гидрофобных частей аминокислот и белков: в воде (В), в структуре льда (Г) и в клатратах (Д).
4. Сочетание в структуре аминокислот протон-донорных и протон-акцепторных групп обеспечивает их участие в водородных связях с другими молекулами, в частности с молекулами растворителя.
Гидрофобность является свойством неполярных молекул и химических групп, она отражает их тенденцию растворяться в неполярных растворителях. В то же время эти неполярные молекулы практически не растворяются в воде и не смешиваются с ней. Среди неорганических веществ гидрофобными считаются благородные газы, среди органических — углеводороды и их производные. Мерой гидрофобности вещества принято считать его коэффициент распределения между органическим растворителем и водой; Тенфорд использовал в качестве такого органического растворителя этанол, Мак-Грегор — 2-бутанол. Это разделило аминокислотные остатки на три сорта: гидрофильные, гидрофобные и те, которые одинаково распределяются между органической и водной фазами. Наиболее полная систематизация гидрофильно-гидрофобных характеристик принадлежит Ганшу, который применял в качестве неполярного растворителя октанол (Leo et al., 1971). Мы использовали из его обзора данные о коэффициентах распределения аминокислот между октанолом и водой для вычисления свободной энергии переноса (G) аминокислоты из воды в октанол. При этом самой гидрофильной аминокислотой является глицин — цвиттерион, не имеющий боковой группы, с собственным дипольным моментом 20 Д.3 Для его перемещения из воды в октанол требуется затратить 17.4 кДж/моль. Все остальные аминокислоты более гидрофобны и перемещаются в неполярное окружение с меньшими затратами энергии. Принимая гидрофобность остатка глицина за 0, можно оценить гидрофобность боковых групп других аминокислот. В табл. 1 представлены расчетные значения свободной энергии перемещения боковой группы аминокислоты из воды в октанол, т. е. из полярного растворителя в неполярный. Эта шкала гидрофобности аминокислотных остатков обнаруживает, что все они в той или иной степени гидрофобны, хотя для большинства из них энергия перемещения в неполярное окружение невелика и соизмерима с тепловой энергией: RT — 2.58 кДж/моль при 37 °С (здесь R — газовая постоянная, Т — температура). В дальнейшем мы будем использовать эту шкалу гидрофобности при оценке межмолекулярных взаимодействий пептидов друг с другом и с другими компонентами клетки — фосфолипидами и нуклеиновыми кислотами, хотя при теоретических расчетах оптимальных конформаций белков наиболее часто используется упрощенная шкала Тенфорда.
Аминокислотные остатки существенно различаются по парциальным мольным объемам в гидратированном состоянии (табл. 1). Самые высокие значения мольных объемов принадлежат самым гидрофобным аминокислотам — тирозину, фенилаланину и триптофану, превосходя значения мольных объемов остатков гидрофильных глутаминовой и аспарагиновой кислот, лизина и аргинина, несущих электрические заряды на боковых группах (Замятнин, 1973).
3 Дебай (Д) — несистемная единица молекулярного дипольного момента: 1 Д = 3.3 — 10-30 К ∙ м.
Таблица 1 Аминокислоты, кодируемые ДНК (по: Leo et al., 1971; Замятнин, 1973)

Измерение гидрофобности органических соединений имеет давнюю традицию, так как фармакологи еще в начале XX в. отмечали корреляцию между гидрофобностью и физиологической активностью токсинов, нейролептиков и наркотических веществ. Предполагалось, что липидные структуры клеточных мембран и миелиновые структуры нервных волокон представляют собой неполярную фазу, куда с высокой селективностью распределяются гидрофобные молекулы. Только позднее Л. Полинг предположил, что в осуществлении физиологических межмолекулярных взаимодействий определяющую роль играет структура гидратационной воды (Pauling, 1961).
В настоящее время экспериментально установлено, что вода — неотъемлемый участник всех биологических процессов на Земле. Структура и подвижность воды составляют основу подвижности всех других компонентов живых систем. Среднее весовое содержание воды в клетках млекопитающих составляет 70 %; на долю белков и фосфолипидов приходится 18 и 3 % соответственно. На все остальные компоненты живой клетки приходится только 9 % ее веса.
Молекула воды имеет геометрию практически равнобедренного треугольника. Расстояния между ядром атома кислорода и протонами равно 0.96 Å. Кристаллографический диаметр молекулы воды равен 2.8 Å. В жидкой воде каждая молекула воды связана с двумя соседними, как это представлено на рис. 1, В—Д. Центральный атом кислорода участвует в четырех водородных связях: в двух из них он донор электронов, в двух других — акцептор. Угол в системе О...Н—О близок к 180°. Такая структура определяет высокую полярность молекулы воды: ее собственный дипольный момент равен 1.84 Д.
Структура молекулы воды и ее межмолекулярные водородные связи играют ключевую роль при гидратации растворенных веществ, в частности пептидов и белков. Вокруг полярных и неполярных групп в воде возникают различные типы гидратационных оболочек. Около неполярных (гидрофобных) групп, не способных к участию в водородных связях, выстраивается ажурная ячейка из молекул воды, связанных только друг с другом, как это проиллюстрировано на рис. 1, Г и Д. Шестичленные изогнутые циклы и ненапряженные плоские пентагоны с углами 108° формируют пространственные многогранники, состоящие из триплетных и более объемных структур с кубической симметрией, — ячейки, внутри которых размещается гидрофобная молекула (Адамсон, 1984). Структуры с таким типом координации молекул воды получили название “клатраты”, т. е. клеточные гидраты (Габуда, 1982), и были относены к классу соединений включения. Эти ажурные рыхлые гидратные структуры имеют среднюю плотность 0.79 г/см3, тогда как плотность обычного льда составляет 0.92 г/см3. Клатраты устойчивы только при наличии в полостях каких-либо молекул или атомов, не способных участвовать в водородных связях, так что количество клатратной воды оказывается максимальным для гидратов гидрофобных веществ (Замятнин, 1973). Не все клатратные структуры воды являются электронейтральными: в пятизвенных циклах дипольные моменты молекул воды направлены наружу по отношению к заключенной в ячейке молекуле (рис. 1, Д).
Для каждого типа таких клатратных соединений имеется критическая температура, выше которой они разрушаются, как бы плавятся, и этот “фазовый” переход наблюдается как эндогенный термический эффект при дифференциальном термическом анализе.
В конце 50-х годов Полинг показал, что вода может формировать комплексные соединения с углеводородами (например, СН4 ∙ 6Н2О) и с углеводородными частями биологически активных молекул, в частности с анальгетиками. Ему принадлежит гипотеза о том, что сочетание полярной (или электрически заряженной) группы с гидрофобными частями молекулы лежит в основе взаимодействия анальгетиков и нейротоксинов с клеточными мембранами (Pauling, 1961).
Если аминокислотный остаток несет электростатический заряд или взаимодействует с молекулами воды как донор или акцептор водородной связи, то вокруг него образуется более плотный слой гидратационной воды, имеющий льдоподобную структуру. Дипольные моменты молекул воды в этом слое ориентированы под действием локального электростатического поля гидратируемой группы (рис. 1, Д). Ионизация карбоксильной или аминогруппы аминокислоты приводит к изменению типа гидратации от клатратного до гидратного, что отображается в системе водородных связей ближайшего окружения. Расчет энергии ассоциации молекул воды с заряженными центрами показал, что знак заряда
частицы влияет на вектор поляризации водного окружения (Русанов, 1978). Радиус сольватации r также зависит от знака и плотности электрического заряда гидратируемой группы в ряду: r+ < r- < r2 < r2+. Например, при конденсации паров воды конденсирующая активность отрицательных ионов на порядок выше активности положительных ионов. По этой причине капли дождя имеют отрицательный избыточный вектор поляризации поверхности, а потенциал атмосферы сохраняет положительное значение.
Изучение динамики гидратационной воды около участков молекулы с разной полярностью позволяет определить границы областей гидратной оболочки, различающихся ориентацией и плотностью упаковки молекул воды (Okouchi et al., 2002).
Ионизация простой органической кислоты (например, уксусной) имеет только один этап депротонирования, который приводит к возникновению отрицательного заряда на карбоксильной группе:
![]()
Равновесие такой реакции определяется единственной константой кислотной диссоциации:
![]()
Кислотная диссоциация (депротонирование) цвиттерионов осуществляется как более сложный, многоступенчатый процесс, при этом константы равновесия на каждом этапе зависят от пути диссоциации всей молекулы. Диссоциация простейшей аминокислоты — глицина — может быть представлена следующей схемой (Чанг, 1980):
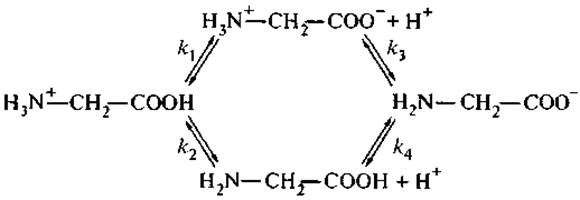
где k1, k2, k3 и k4 — микроскопические константы диссоциации, связанные термодинамическим соотношением:
k1k3 = k2k4.
Однако экспериментально можно определить только две константы ионизации: К1 = k1 + k2 — для карбоксильных групп; К2 = k3k4/k3 + k4 — для аминогрупп.
Расчеты, выполненные на основе данных потенциометрического титрования глицина, показывают, что в зависимости от пути ионизации молекулы (изменение pH от кислых значений к щелочным или наоборот) микроскопические константы диссоциации аминогруппы (k2 и k1) различаются на два порядка. Изменение ионизации той или иной группы влияет на взаимодействие этой группы с ближайшим окружением, меняя соотношение клатратной и гидратной структур в связанной воде.
Физико-химические свойства аминокислот и их полифункциональность важны для осуществления многочисленных регуляторных функций этих веществ в живых организмах. Индивидуальные аминокислоты, их производные (пептиды) и продукты метаболизма (аммиак, мочевина, ароматические амины) служат, в частности, медиаторами нервных окончаний, сигналами связи с внешней средой, ингибиторами отдельных биохимических реакций, являются адаптогенами и основой для синтеза нейропептидов и гормонов (Кричевская и др., 1983). Плазма крови представляет собой их депо и обеспечивает транспорт аминокислот к определенным органам.
Фонд свободных аминокислот в клетках живых организмов имеет эволюционную, органную и тканевую специфичность. Например, аминокислотный состав мозга существенно отличается от состава других органов и тканей присутствием избыточного количества дикарбоновых кислот и их амидов: они составляют две трети от общего количества аминокислот в мозге всех видов животных. Глутамин, аспарагин и их остатки в составе пептидов в организме неферментативно гидролизуются до соответствующих дикарбоновых кислот. В связи с этим важно отметить, что белки молодых клеток характеризуются более высокой степенью амидированности, чем белки стареющих клеток (Пушкина, 1977).
Четыре ароматические аминокислоты — гистидин, тирозин, фенилаланин и триптофан обладают повышенной химической активностью боковых групп. Она определяется системой сопряженных связей и делокализованных электронов и способностью этих групп участвовать в реакциях нуклеофильных и электрофильных замещений. Ароматические аминокислоты составляют основу многих биологически активных производных: гормонов, медиаторов, коферментов.
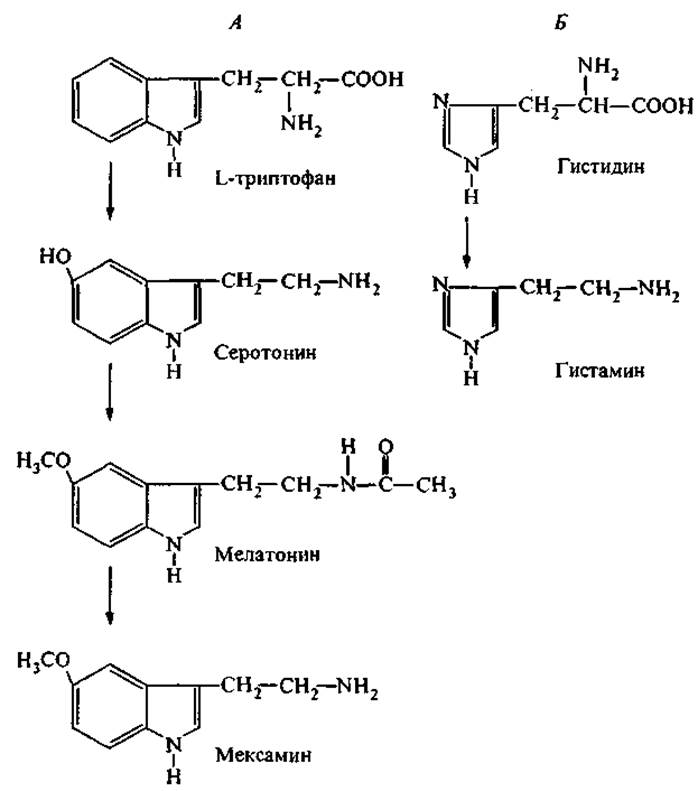
Рис. 2. Пути трансформации аминокислот в молекулярные регуляторы биологических функций.
А — преобразование триптофана: серотонин — нейромедиатор, возбуждающий пост-ганглионарные нервные волокна; мелатонин — гормон эпифиза; мексамин — радиопротектор. Б — преобразование гистидина в гистамин — биологически активный амин, обладающий гормональным действием и медиаторными функциями.
На рис. 2 представлены примеры таких превращений триптофана и гистидина.
Триптофан является незаменимой аминокислотой и в природе синтезируется микроорганизмами. Суточная потребность человека в триптофане составляет 250 мг, а недостаток триптофана переносится тяжелее, чем голод. Собственно триптофан обладает широким спектром физиологической активности и положительно влияет на липидный обмен и на синтез белков в печени. В малых дозах триптофан обнаруживает гипогликемический эффект, но при больших дозах этот эффект меняется на противоположный. Например, диабетогенное действие триптофана наблюдалось на крысах при скармливании дозы 2.5 г на 1 кг массы тела, что в 700 раз выше нормы суточного потребления (Рудзит, 1981). В дальнейшем мы увидим, что несколько пептидных гормонов желудочно-кишечного тракта (ЖКТ), стимулирующих выделение инсулина, содержат триптофан в своих активных детерминантах (Приложение, табл. II). Можно предполагать, что свободный триптофан в сверхвысоких дозах выступает конкурентом (антагонистом) этих гормонов и ингибирует специфические рецепторы на поверхности клеток, секретирующих инсулин.
Гистидин также является незаменимой аминокислотой. Его регуляторные функции определяются химическими свойствами боковой группы — имидазола. В частности, эта группа участвует в окислительно-восстановительных реакциях и способна устанавливать координационные связи с переходными металлами. В свободном состоянии гистидин содержится в тканях в очень низкой концентрации. В то же время он входит в каталитические (активные) центры многих ферментов (рибонуклеаза, химотрипсин, конвертаза) и регуляторных пептидов (карнозин, гистатин, нейрокинины) благодаря донорно-акцепторным свойствам своей имидазольной группы. Декарбоксилирование гистидина приводит к образованию гистамина — медиатора, который регулирует сосудистое давление, проницаемость капилляров и аллергические реакции. Как медиатор гистамин имеет три вида клеточных рецепторов, в том числе в клетках головного мозга.
Иначе говоря, диапазон регуляторных функций аминокислот и их производных обеспечивается их физико-химической полифункциональностью и участием в обратимых биохимических реакциях.
Как уже указывалось, аминокислоты в водной среде являются биполярными ионами, и образование между ними пептидной связи в водных растворах оказывается термодинамически невыгодным процессом. Синтез пептидов традиционно проводят в органических растворителях после предварительной защиты тех групп, которые не участвуют в образовании пептидной связи. В зависимости от длины и компонентного состава полипептида выбирается оптимальная стратегия его синтеза: жидкофазный, твердофазный или рекомбинантный (Andersson et al., 2000).
Образование пептидной связи может быть представлено следующей схемой:
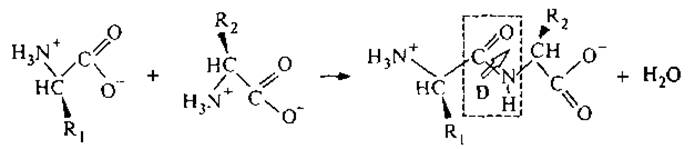
В полученном дипептиде свободная электронная пара азота сопряжена с двойной связью карбонильной группы, так что связь С—N отчасти сдвоена и вращение вокруг нее затруднено. Пептидная связь имеет постоянный дипольный момент D. Боковые группы R1 и R2 определяют комбинацию физико-химических свойств, присущих исходным индивидуальным аминокислотам. Как известно, при всех превращениях, если не происходит разрыва связи у асимметричного атома, конфигурация молекулы сохраняется. Поэтому при образовании пептидной связи сохраняются основные свойства исходных аминокислот: 1) оптическая активность, обусловленная хиральностью строения аминокислотных остатков; 2) способность участвовать в межмолекулярных водородных связях.
Однако при объединении аминокислот в молекулу пептида для каждой частной аминокислотной последовательности складывается индивидуальное соотношение гидрофильности и гидрофобности боковых групп пептида (Alberts et al., 1994).
Пространственное распределение разноименных электрических зарядов и дипольный момент пептидной связи, равный 3.5 Д, определяют постоянный дипольный момент и высокую поляризуемость пептида. Благодаря этим приобретенным свойствам дипептиды имеют более широкий спектр энергетических состояний, чем индивидуальные аминокислоты, что, однако, компенсируется уменьшением пространственных степеней свободы системы. Каждая из простых молекул имеет в растворе 6 степеней свободы движения: 3 вращательные и 3 поступательные. Две не взаимодействующие друг с другом аминокислоты имеют 12 степеней свободы; при их объединении в один дипептид число степеней свободы уменьшается до 6. С точки зрения статистической термодинамики, это равнозначно увеличению порядка в системе и соответствующему уменьшению энтропии.
Следует отдельно остановиться на принципиально новом качестве, которое приобретают аминокислоты, объединяясь в полипептидную цепь. Это качество — комплементарность подвижных конформаций.
Общее понятие комплементарности (дополнительности) относится к категориям ранних натурфилософских представлений. В эпоху развития алхимии сформировались два описательных представления: “подобное к подобному” и “противоположности сближаются”. Первое опиралось на опыт разделения и очистки веществ, второе — на опыт химических превращений, при которых элементы с противоположными качествами взаимодействуют и дополняют друг друга. На современном уровне представлений о строении веществ принцип “подобное к подобному” воплотился в теорию гидрофильно-гидрофобных взаимодействий, а комплементарность иллюстрируется как притяжение положительных и отрицательных зарядов (в электростатике), как совмещение выпукло-вогнутых поверхностей, как взаимное соответствие элементов объекта и его зеркального отражения (матричная комплементарность ранней полиграфии). Во всех этих примерах “противоположности” рассматриваются как неизменные объекты.
Биологические макромолекулы, в частности пептиды, сохраняют комплементарность межмолекулярных взаимодействий в достаточно широком диапазоне конформаций. Эта их способность лежит в основе каталитической активности ферментов, и модель комплементарного взаимодействия “ключ—замок” была впервые использована на заре энзимологии. Позднее эта же модель использовалась при обсуждении специфичности связывания антигена с антителом и селективности взаимодействия рецептора с лигандом.
При исследовании нуклеиновых кислот была впервые использована модель матричной комплементарности нуклеотидных пар. С ее помощью было доказано, что эти макромолекулы управляют точным копированием собственной структуры, так как исходная макромолекула является матрицей для образования следующей.
Принцип матричной комплементарности очень прост и элегантен. Он состоит в том, что каждый элемент упорядоченной молекулярной конструкции (электростатический заряд, протон-донорная или гидрофобная группа) в том же порядке копируется на другую макромолекулу, элементы которой комплементарны элементам первой молекулы.
При ближайшем рассмотрении матричная комплементарность была обнаружена и при синтезе пептидов, и при формировании фосфолипидных бислойных мембран, и при самосборке полимеров (Conjugated oligomers..., 1998).