Фізіологія людини - Вільям Ф. Ґанонґ 2002
Ендокринна система, метаболізм і репродуктивна функція
Ендокринні функції підшлункової залози й регулювання метаболізму вуглеводів
Регулювання секреції інсуліну
Нормальна концентрація інсуліну, виміряна радіоімунологічними дослідженнями в периферійній венозній крові, у здорової людини натще становить 0-70 мкОД/мл (0- 502 пмоль/л). Рівень базальної секреції інсуліну - 1 ОД/г. Приймання їжі супроводжується 5-10-разовим підвищенням рівня інсуліну. Отже, загальна денна кількість у нормальної людини становить близько 40 ОД (287 нмоль).
Вплив глікемії
Секреція інсуліну головно контрольована за допомогою впливу глікемії на В-клітини підшлункової залози завдяки' механізму зворотного зв’язку. Глюкоза потрапляє в В-клітини через транспортер GLUT 2, який є у значній кількості і не залежить від інсулінового активування. Глюкозу метаболізує ензим глюкокіназа, що є обмежувальною ланкою у реакції. Утворюється АТФ, що закриває АТФ-чутливі К+-канали (рис. 19-14). Унаслідок цього зменшений витік К+ деполяризує клітинну мембрану. Це приводить до відкривання потенціалозалежних Са2+-каналів, і Са2+ надходить у клітину. Збільшення внутрішньоклітинного кальцію веде до вивільнення інсуліну шляхом екзоцитозу.
Зв’язок вивільнення інсуліну з рівнем глюкози в плазмі показаний на рис. 19-13. Маноза також стимулює секрецію інсуліну (табл. 19-6). Таку ж дію має фруктоза, яка внутрішньоклітинно перетворюється в глюкозу. Маногептулоза і 2-дезоксиглюкоза, які оберігають глюкозу від метаболізації, пригнічують секрецію інсуліну.
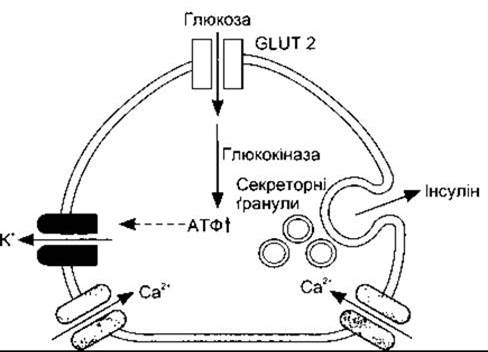
Рис. 19-14. Регулювання секреції інсуліну глікемією. Глюкоза надходить у В-клітини через транспортер глюкози GLUT 2, її метаболізує глікокіназа, збільшуючи вміст внутрішньоклітинного АТФ. АТФ інгібує АТФ-чутливі К+-канали, зменшуючи відтікання К+ і деполяризуючи клітину. Це активує потенціалозалежні Са2+-канали, унаслідок чого збільшується надходження Са2+ у клітину, що спричинює вивільнення інсуліну шляхом екзоцитозу.
В експериментальних тварин (рис. 19-15) та людей вплив глюкози на секрецію інсуліну двофазовий: швидке збільшення секреції з наступною повільною пролонгованою відповіддю. Первинна реакція виникає внаслідок вивільнення утвореного інсуліну у відповідь на збільшення цитоплазматичного Са2+. Пролонгована реакція є наслідком вивільнення новосинтезованого інсуліну, аж доки не відбудеться блокування інгібіторами білкового синтезу. Якщо перфузія глюкози відновлюється після періоду спокою, то відповідь на глюкозу збільшується.
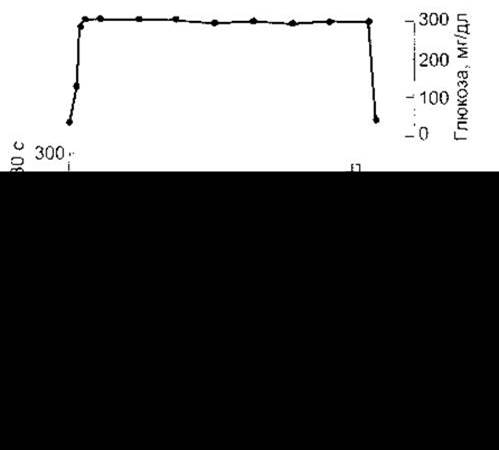
Рис. 19-15. Секреція інсуліну з перфузійної підшлункової залози щура у відповідь на тривале введення глюкози. Наведено середні значення трьох препаратів Верхній запис відображає концентрацію глюкози у перфузійній суміші (відтворено за дозволом з Curry DL, Bennett LL, Grodsky GM: Dynamics of insulin secretion by the perfussed rat pancreas. Endocrinology 1968;83:572).
Таблиця 19-6. Фактори, що впливають на секрецію інсуліну
Стимулятори |
Інгібітори |
Глюкоза |
Соматостатин |
Маноза |
2-Деокси глюкоза |
Амінокислоти (лейцин, аргінін та ін.) |
Маногептулоза |
Кишкові гормони (ШІП, GLP-1 [7-36], гастрин, секретин, ХЦК; інші?) |
а-Адренергічні стимулятори (норадреналін, адреналін) |
ß-Кетокислоти |
а-Адренергічні блокатор(анаприлін) |
Ацетилхолін |
Галангін |
Глюкагон |
Діазоксид |
Циклічний АМФ та різні цАМФ-утворювальні сполуки |
Тіазидні сечогінні |
ß-Адренергічні стимулятори |
Виснаження запасів К+ |
Дифенін |
|
Теофілін |
Апоксан |
Сульфонілсечовини |
Інгібітори мікротрубочок |
Інсулін |
Механізми зворотного контролювання глюкози плазми чи секреції інсуліну мають високу точність, адже рівні глюкози й інсуліну змінюються паралельно з високим ступенем відповідності.
Похідні білків та жирів
Інсулін стимулює входження амінокислот у білки і запобігає катаболізму жирів з утворенням ß-кетокислот. Тому й не дивно, що аргінін, лейцин та деякі інші амінокислоти, а також ß-кетокислоти та ацетоацетат стимулюють секрецію інсуліну. Як і глюкоза, ці сполуки внаслідок обміну утворюють АТФ, який закриває АТФ-чутливі К+-канали у В-клітинах. Окрім цього, L-аргінін є попередником NO, а NО стимулює секрецію інсуліну.
Пероральні гіпоглікемічні засоби
Толбутамід та інші похідні сульфонілсечовини, такі як ацетогексамід, толазамід, гліпізид та глібурид, є пероральними активними гіпоглікемічними засобами, що знижують рівень глюкози в крові, підвищуючи секрецію інсуліну. Вони діють лише в пацієнтів з деяким залишком В-клітин, неефективні після гіанкреатектомії чи в разі цукрового діабету першого типу; зв’язуються з АТФ-інгібованими К+- каналами в мембранах В-клітин, пригнічують активність каналів, деполяризуючи мембрану В-клітин і збільшуючи надходження Са2+, а, отже, і вивільнення інсуліну.
Тривала гіперінсулінемічна гіпоглікемія новонароджених - це рідкісний стан, за якого кількість інсуліну збільшується, незважаючи на гіпоглікемію. Цей стан спричинений інактивувальною мутацією АТФ-інгібованих К+-каналів. Лікування полягає у введенні діазоксиду - препарату, що збільшує активність К+-каналів, або, у легших випадках - у субтотальній панкреатектомії.
Бігуанідини, фенформін та метформін є пероральними гіпоглікемічними засобами, що діють без інсуліну. Фенформін спричинив лактат-ацидоз у надто великої кількості пацієнтів, і внаслідок серйозності цього побічного ефекту його вилучено з ринку Сполучених Штатів Америки. Метформін також може зумовити лактат-ацидоз, однак такі випадки становлять лише 5-10% від випадків лактат-ацидозу, пов’язаного з фенформіном. Первинна дія метформіну пов’язана зі зменшенням глюконеогенезу, а, отже, зменшенням печінкового виходу глюкози. Його інколи поєднують із препаратами сульфонілсечовини у разі лікування діабету другого типу (див. нижче).
Троглітазон (резулін) та близькі тіазолідиндіони також використовують для лікування діабету, оскільки вони збільшують інсулінопосередкований периферійний глюкозний розподіл, зменшуючи в такий спосіб інсулінову резистентність, а також зв’язуються й активують пероксисомний проліфератор-активований рецептор у (PPARy - від англ. peroxisome proliferator-activated receptor у) у ядрі клітин (див. Розділ 1). Активування цього рецептора, який є членом надродини гормоночутливих ядерних факторів транскрипції (див. Розділ 1), має виняткову здатність нормалізувати численні метаболічні функції.
Циклічний АМФ і секреція інсуліну
Фактори, що збільшують рівні цАМФ у В-клітинах, посилюють секрецію інсуліну, можливо, шляхом збільшення внутрішньоклітинного Са2+. Вони містять ß-адренергічні агоністи, глюкагон та фосфодіестеразні інгібітори, такі як теофілін.
Катехоламіни чинять подвійний вплив на секрецію інсуліну: вони блокують її через а2-адренергічні рецептори та стимулюють через ß-адренергічні рецептори. Сумарним ефектом впливу адреналіну та норадреналіну є пригнічення (секреції інсуліну). Однак якщо катехоламіни вводять після приймання а-адренергічних блокувальних препаратів, то сповільнення секреції переходить у її стимулювання.
Вплив автономної нервової системи
Відгалуження правого блукаючого нерва іннервують підшлункові острівці, а його стимулювання зумовлює збільшення секреції інсуліну через рецептори М4 (див. табл. 4-2). Атропін блокує цю відповідь, а ацетилхолін стимулює секрецію інсуліну. Ацетилхолін, як і глюкоза, впливає внаслідок збільшення цитоплазматичного Са2+, однак ацетилхолін активує фосфоліпазу С з вивільненням ІФ3, що забезпечує вихід Са2+ з ендоплазматичної сітки.
Стимулювання симпатичних нервів підшлункової залози пригнічує секрецію інсуліну. Це відбувається шляхом вивільнення норадреналіну, який діє на а2-адренергічні рецептори. Та якщо а-адренергічні рецептори блоковані, то стимулювання нервів підшлункової залози спричинює збільшення секреції інсуліну, що зумовлена р2-адренергічними рецепторами. Поліпептид галанін, знайдений у деяких нервах автономної нервової системи, що іннервують острівці, сповільнює секрецію інсуліну, активуючи К+-ка- нали, інгібовані АТФ. Отже, автономна іннервація підшлункової залози задіяна в регулюванні секреції інсуліну. Ефекти глюкози не потребують інтактної іннервації, хоч вони і відбуваються в трансплантованій підшлунковій залозі; однак є докази, що нервові волокна підтримують нормальну чутливість острівців до глюкози.
Кишкові гормони
Глюкоза, яку вводять перорально, зумовлює більший інсуліностимулювальний ефект, ніж уведена внутрішньовенно; а амінокислоти, введені перорально, також спричинюють сильнішу інсулінову відповідь, ніж внутрішньовенні. Ці спостереження спонукали дослідити можливість того, що речовини, які секретує слизова оболонка шлунково-кишкового тракту, стимулюють секрецію інсуліну. Глюкагон, його похідні, секретин, холецистокінін (ХЦК), гастрин та шлунковий інгібіторний поліпептид (ШІП) мають таку дію (див. Розділ 26), а ХЦК уможливлює інсуліностимулювальний ефект амінокислот. Однак ШІП-єдиний з цих пептидів, що спричинює стимулювання, якщо його вжито в дозах, що роблять рівні ШІП у крові сумірними з рівнями, які створює пероральна глюкоза.
Останнім часом увага звернута на глюкагоноподібний пептид 1 (7-36) (GLP-1 [7-36], від англ. glucagon-like polypeptide 1 (7-36)) як додатковий кишковий фактор, що стимулює секрецію інсуліну. Цей поліпептид є продуктом препроглюкагону (див. нижче). У В-клітинах наявні рецептори GLP-1 (7-36) і ШІП; GLP-1 (7-36) - це сильніший інсулінотропний гормон, ніж ШІП. Обидва - ШІП та GLP- 1 (7-36) - діють, збільшуючи надходження Ca2+ через потенціалозалежні Са2+-канали.
Можлива роль підшлункового соматостатину й глюкагону в регулюванні секреції інсуліну розглянута нижче.
Вплив виснаження К+
Виснаження пулу йонів калію зменшує секрецію інсуліну, і в пацієнтів за таких станів, наприклад із первинним гіперальдостеронізмом (див. Розділ 20), розвиваються зміни кривої діабетичної глюкозної толерантності. За насичення К+ можливе повернення до норми. Тіазидні діуретики, що спричинюють втрату К+ та Na+ із сечею (див. Розділ 38), знижують толерантність до глюкози і погіршують перебіг діабету. Вони виявлять таку дію, найперше, внаслідок ефектів виснаження К+, хоча деякі з них також зумовлюють ушкодження клітин острівців.
Довготривалі зміни у відповіді В-клітин
Інсулінова відповідь на конкретний стимул частково визначена секреторною історією В-клітин. Особи, що споживали їжу з високим вмістом вуглеводів протягом кількох тижнів, не лише мають вищі рівні інсуліну в плазмі (натще), а й виявляють на введення глюкози більш секреторну відповідь, ніж особи, що споживали ізокалорійну їжу з низьким вмістом вуглеводів.
Хоча В-клітини відповідають на стимулювання гіпертрофією, як і інші ендокринні клітини, вони виснажуються й припиняють секрецію (виснаження В-клітин), коли стимулювання є сильним чи тривалим. Підшлунковий резерв великий, тому досить важко спричинити виснаження в нормальних тварин; однак якщо підшлунковий резерв зменшується частковою панкреатектомією чи малими дозами алоксану, то виснаження В-клітин, що залишились, може бути зумовлене будь-яким процесом, що хронічно підвищує рівень глюкози в плазмі. Це є причиною діабету, що виникає у тварин з обмеженими підшлунковими запасами у разі дії екстрактів передньої частки гіпофізу, гормону росту, тиреоїдних гормонів чи тривалого введення самої глюкози. Спричинені гормонами діабети у тварин є спочатку зворотними, проте в разі тривалої дії стають постійними. Їх, зазвичай, називають за агентом, який їх утворює, наприклад, гіпофізарний діабет, тиреоїдний діабет. Довготривалі діабети, що зберігаються після дії препаратів, позначають префіксом мета-, наприклад, метагіпофізарний діабет чи метатиреоїдний діабет. Якщо ж інсулін вживали разом з діабетогенними гормонами, то В-клітини є захищеними, можливо, внаслідок зниження рівня глюкози в плазмі, і тому діабет не розвивається.
Цікаво, що в разі контролювання резерву В-клітин можуть бути задіяні генетичні фактори. У мишей нокаутом гена IRS-1 (див. вище) є сильна компенсаторна відповідь клітин В. Проте у випадку нокауту гена IRS-2 компенсація зменшена, і утворюється серйозніший діабетичний фенотип.