Основы биоинформатики - Огурцов А.Н. 2013
Методы биоинформационного анализа
Филогенетический анализ
Кладистические методы
В соответствии с кладистическим подходом в группы объединяют только те виды, которые имеют общие приобретённые признаки, то есть признаки, которые отсутствовали у их дальних предков.
Кладистический подход, как полагают, является наилучшим методом для филогенетического анализа, потому что он принимает текущую эволюционную теорию и опирается на нее, то есть предполагает, что новые виды образуются при разветвлении эволюционных линий (то есть путём кладогенеза).
Кладом называют монофилетический таксон.
Клад - это группа организмов или генов, в которую входит ближайший общий предок всех её членов и все потомки этого ближайшего общего предка. Термин "клад" (англ. clade) произошёл от греческого слова κλаδоζ, означающего "ветвь", "ответвление" или "прут". Изредка в литературе этот термин встречается в женском роде - "клада".
Таксоном называют систематическую группу в классификации организмов. Узел представляет собой точку разветвления эволюционной линии. В некоторых случаях длины ветвей соответствуют степени расхождения видов (рисунок 65).

Рисунок 65 - Родство трёх млекопитающих, представленное в виде ветви филогенетического дерева
Примеры кладограмм, в которых порядок разветвлений соответствует эволюционным последовательностям видообразований у предков, представлен на рисунках 21 и 22.
Кладистический подход рассматривает возможные направления эволюции, выводит характеристики предков на всех узлах и выбирает оптимальное дерево согласно некоторой эволюционной модели. Отправной точкой кладистики служит тот факт, что члены одной группы, или одного клада, имеют общую эволюционную историю и более тесно связаны друг с другом, чем с членами любой другой группы.
Данная группа определяется по некоторым общим характерным особенностям, которые отсутствовали у далёких предков. Такими общими приобретёнными признаками могут быть любые характеристики, которые можно визуально отметить и описать. Обычно кладистический анализ проводят или по набору фенотипических признаков, или по множеству пар оснований или аминокислот в последовательности.
В кладистике приняты три основных допущения.
1. Организмы любой группы связаны между собой происхождением от общего предка.
2. Эволюционные линии периодически разветвляются.
3. С течением времени у потомков происходит изменение характеристик.
Кладистические методы имеют дело исключительно с паттернами наследования, полученными из анализа возможных деревьев таксонов. Они нацелены на выбор правильного дерева и используют детальные модели эволюционных процессов.
Наиболее популярными кладистическими методами являются:
1) метод молекулярной филогении - метод максимальной экономии (Maximum Parsimony);
2) метод наибольшего правдоподобия (Maximum Likelihood).
Эти методы ориентированы на данные о последовательностях и начинаются с множественного выравнивания. Оба метода не ориентируются на анатомические особенности организмов, такие как средний вес взрослой особи.
Метод наибольшей экономии (Maximum Parsimony или метод минимальной эволюции), предложенный Фитчем (W. Fitch), предсказывает эволюционное дерево, которое минимизирует число шагов, необходимых для воспроизведения наблюдаемого изменения в последовательностях.
Для того чтобы с наибольшей вероятностью предсказать, в каких позициях последовательностей символы (элементы последовательностей) совпадают, требуется построить множественное выравнивание этих последовательностей. В таких позициях множественного выравнивания будут расположены вертикальные столбцы с совпадающими элементами последовательностей. Для каждой выровненной позиции определяют такие филогенетические деревья, которые требуют наименьшее число эволюционных изменений, необходимых для того, чтобы произвести наблюдаемые изменения в последовательностях. Такой анализ проводят для каждой позиции в выравнивании последовательностей. Наконец, определяют те деревья, которые производят наименьшее число изменений в целом для всех позиций последовательностей.
Метод максимальной экономичности применяют для построения деревьев на основе минимального числа мутаций, необходимых для преобразования одной последовательности в другую.
Например, пусть даны виды, в которых есть гомологичные последовательности ATCG, ATGG, ТТСА, TCGA (рисунок 66).
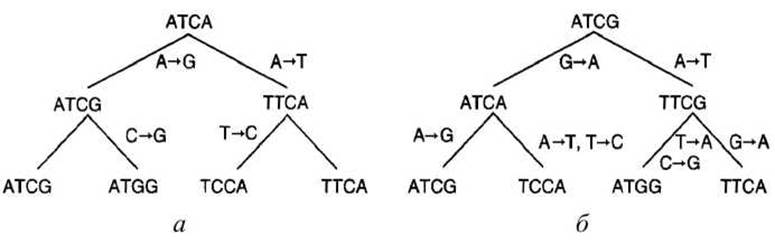
Рисунок 66 - Два генеалогических дерева мутаций
Дерево, представленное на рисунке 66(a), постулирует 4 мутации. Альтернативное дерево, представленное на рисунке 66(6), постулирует 7 мутаций. Отметим, что второе дерево постулирует, что замена G→А в четвертой позиции происходит независимо дважды.
Дерево (а) является оптимальным в соответствии с методом максимальной экономии, поскольку никакое другое дерево не даёт меньшего количества мутаций.
Во многих случаях несколько деревьев могут давать одинаковое количество мутаций, меньшее, чем другие деревья. В этих случаях метод максимальной экономии не даёт единственного ответа.
Программное обеспечение для построения филогенетических деревьев - пакет программ PHYLIP (PHYLogeny Inference Package) - состоит из 35 программ, которые не имеют графического интерфейса:
http://cmgm.stanford.edu/phylip/;
http://evolution.genetics.washington.edu/phylip.html.
Входящие данные представлены в собственном формате PHYLIP. Файл outtree, содержащий дерево, представлен в универсальном Ньюик-формате (Newick tree format, или Newick notation, или New Hampshire tree format).
К основным средствам анализа по методу максимальной экономичности, включенным в пакет PHYLIP, относятся программы DNAPARS, DNAPENNY, DNACOMP, DNAMOVE и PROTPARS.
Метод наибольшего правдоподобия (Maximum Likelihood method), основанный на вероятностном расчёте, позволяет найти дерево, которое наилучшим образом описывает изменения, отмеченные в наборе последовательностей. Этот метод подобен методу максимальной экономичности в том, что анализ проводят для каждого столбца множественного выравнивания последовательностей. При этом учитывают все возможные деревья.
По каждому построенному дереву оценивают число вероятных изменений, или мутаций, вызвавших наблюдаемые изменения в последовательностях. Поскольку частота появления новых мутаций очень мала, постольку чем больше мутаций необходимо для приведения некоторого дерева в соответствие с имеющимися данными, тем менее правдоподобно это дерево. Для каждой топологии дерева варьируется скорость эволюции и оптимизируются параметры для того, чтобы максимизировать правдоподобие порождения наблюдаемых последовательностей.
Деревья с наименьшим количеством изменений будут наиболее правдоподобны.
Метод максимального правдоподобия позволяет построить ожидаемую модель изменений последовательности и для всех остатков взвесить вероятности их замен на любые другие остатки. Пакет PHYLIP включает в себя две программы - DNAML и DNAMLK, - предназначенные для анализа филогенетических отношений по методу максимального правдоподобия.
Проблема переменной скорости эволюции. Предположим, что четыре вида А, В, С, D имеют филогенетическое дерево, показанное на рисунке 67(a). Это дерево соответствует матрице расстояний, представленной на рисунке 68(a).
Предположим, что вид D эволюционирует очень быстро, хотя дерево не меняется. Тогда наблюдается матрица расстояний, показанная на рисунке 68(6). Этой матрице соответствует некорректное филогенетическое дерево, показанное на рисунке 67(6).
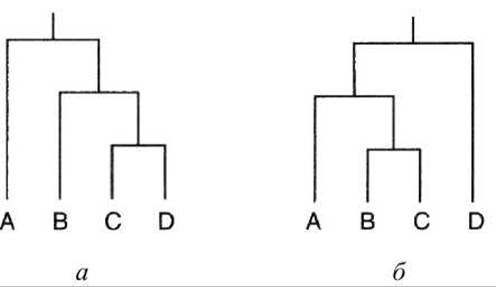
Рисунок 67 - Два филогенетических дерева мутаций
Все обсужденные выше методы не застрахованы от ошибок подобного рода, если скорость эволюции сильно варьирует на разных ветвях дерева.

Рисунок 68 - Матрицы расстояний, соответствующие филогенетическим деревьям рисунка 67
Чтобы проверить есть ли такие вариации, следует принять в рассмотрение внешнюю группу (outgroup) — виды, которые заведомо более удалены от всех видов, для которых строится дерево.
Например, если скорость эволюции у приматов постоянная, то мы ожидаем увидеть примерно одинаковые расстояния между приматами и, скажем, коровой. Если это не так, то неверно предположение о постоянстве скорости эволюции приматов.
Вычислительный анализ. Кладистические методы (максимальной экономии и наибольшего правдоподобия) более точны, чем простые методы кластеризации, такие как UPGMA, но требуют намного больше вычислительных ресурсов для решения разумных задач.
Полное количество возможных деревьев, которое должно быть просмотрено кладистическими методами, очень быстро растёт с увеличением количества видов. В результате во многих интересных случаях эти методы могут дать только приближённое решение, даже при существенных предположениях.
Поскольку вычисление филогении зачастую приближённое, то важно проверить их. Методы проверки включают:
1. Сравнение филогении полученных из разных характеристик таксонов. Являются ли они согласованными? Если деревья, полученные из разных характеристик имеют согласованные поддеревья, то они, по-видимому правильные, в то время как несогласованные поддеревья неправильные.
2. Анализ подмножеств таксонов может дать тот же ответ по отношению к подмножеству - поддерево, построенное на подмножестве должно соответствовать полному дереву.
3. Формальные статистические тесты, включающие в себя пересчёт на подмножестве исходных данных. Эти методы называются jackknife и bootstrap.
4. Если дерево имеет очень длинные ветви, то имеются серьёзные основания предполагать, что мы имеем неравномерность эволюции, а в этом случае необходимо использовать внешнюю группу.
Jackknife проводит вычисления на случайном подмножестве данных. Для построения филогении по множественному выравниванию отбираются случайные наборы позиций выравнивания и для них проводятся вычисления независимо. Если при этом восстанавливаются одинаковые поддеревья, то дерево признаётся правильным, а в противном случае - неправильным.
Bootstrap работает аналогично, но только случайно отобранные позиции могут появляться в выборке несколько раз так, чтобы размер выборки совпадал с исходной выборкой.