БИОТЕХНОЛОГИЯ РАСТЕНИЙ И БИОБЕЗОПАСНОСТЬ - А. П. Ермишин - 2015
ГЛАВА 9. ОСНОВЫ ГЕНЕТИЧЕСКОЙ ИНЖЕНЕРИИ РАСТЕНИЙ
Существует много определений генетической инженерии. На взгляд авторов, суть этой новой технологии можно выразить следующим образом. Генетическая инженерия - это технология получения новых комбинаций генетического материала путем проводимых вне клетки манипуляций с молекулами нуклеиновых кислот и переноса созданных конструкций генов в живой организм, в результате которого достигается их включение и активность в этом организме и у его потомства.
Исходя из этого определения, процесс создания генетически модифицированных организмов (далее - ГМО) можно разделить на несколько этапов. Первый этап включает выделение и идентификацию отдельных генов (соответствующих фрагментов ДНК или РНК), которые собираются перенести другим организмам, а также соответствующих регуляторных элементов. Иногда гены или их части синтезируют искусственно. Затем эти гены и регуляторные элементы соединяют между собой в определенном порядке с помощью чисто химических методов (технология рекомбинантных ДНК, или генная инженерия). Все названные манипуляции проводят вне организма in vitro. В результате получается генетическая конструкция, которая содержит один или несколько генов (точнее, фрагментов ДНК, которые кодируют последовательность аминокислот протеинов - продуктов генов), а также все необходимые регуляторные элементы, обеспечивающие активность этих генов (трансгенов) после их переноса в организмы.
Технологию получения рекомбинантных молекул ДНК по праву считают центральным звеном генетической инженерии. Можно сказать, что годом рождения генетической инженерии является 1972 г., когда появились первые публикации сотрудников лаборатории П. Берга (США). В них сообщалось о получении кольцевой молекулы ДНК вируса SV40 путем последовательного ее разрезания рестриктазой RI и сшивания ДНК лигазой (J. Mertz, R. Davis, 1972), а также возможности с помощью этих ферментов встраивать в такую молекулу гены других организмов: фага-λ и галактозный оперон бактерии Е. coli (D. Jackson, R. Symons, P. Berg, 1972). В 1980 г. П. Берг за эти работы был удостоен Нобелевской премии в области химии.
Следующий этап создания ГМО - перенос трансгенов в отдельные живые клетки (процесс трансформации или, как принято его называть в последнее время, - генетической модификации), где они могут реплицироваться и передаваться дочерним клеткам, образовавшимся при делении трансформированных клеток. Для этого полученные in vitroгенетические конструкции соединяют с ДНК так называемого вектора, в качестве которого чаще всего используют плазмиды.
Для одноклеточных организмов процесс генетической модификации заканчивается, как правило, внедрением в них рекомбинантной плазмиды и последующим отбором трансформированных клеток. Лишь в отдельных случаях для более высокой стабильности трансформантов добиваются включения трансгенов в бактериальную хромосому. В случае же высших многоклеточных организмов встраивание трансгенов в генетический материал клетки (ДНК хромосом или клеточных органелл - хлоропластов, митохондрий) является обязательным. Основной метод переноса трансгенов в геном растений - агробактериальная трансформация, природный механизм горизонтального переноса генов, приспособленный для нужд генетической инженерии.
Благодаря свойству тотипотентности из трансформированных растительных клеток после отбора на селективных средах (в генетических конструкциях помимо целевого гена, как правило, также имеется селективный ген) можно восстановить целый организм. Биосинтез новых для организма протеинов, которые кодируются привнесенными генами, выступает в качестве основы для проявления у него нового селекционного признака, например, толерантности к гербицидам, антибиотикам, устойчивости к насекомым-вредителям и т. д.
Рассмотрим подробнее, каким образом осуществляют перечисленные манипуляции.
9.1. Получение рекомбинантных ДНК
Технологию рекомбинантных ДНК можно использовать для манипуляций с фрагментами ДНК, выделенными из живых организмов или синтезированными искусственно. Эта технология позволяет также вносить определенные изменения в структуру естественных генов и их регуляторных элементов, комбинировать их в произвольной последовательности. Для того чтобы проводить названные манипуляции, необходимо иметь возможность разрезать молекулы ДНК на отдельные определенные фрагменты, а затем сшивать их в нужном сочетании. Для реализации первой из названных задач используют наборы ферментов - эндонуклеаз рестрикции (рестриктаз).
Первые рестриктазы, пригодные для генно-инженерных целей, были выделены в 1970 г. Г. Смитом (США), обнаружившим, что бактерия Haemophillus influenzae быстро расщепляет ДНК фагов. В ходе исследований, проведенных им, было показано, что нуклеазная активность (способность разрезать молекулы нуклеиновых кислот ДНК и РНК), характерная для живых бактерий, сохраняется и в бесклеточном экстракте, полученном из них. Выяснилось, что нуклеазная активность обусловлена присутствием в экстракте фермента-рестриктазы, получившего название HindII. Было установлено, что выделенный и очищенный фермент способен узнавать последовательности нуклеотидов
ГТПи ↓ ПуАЦ
ЦАПу ↑ ПиТГ,
где Пи - любое пиримидиновое основание, а Пу - пуриновое основание (в англоязычной литературе используют символы Y и R соответственно) и проводить расщепление молекулы ДНК (место расщепления указано стрелками). Интересно, что рестриктаза HindII способна разрезать ДНК из различных источников, в том числе E. coli, но при этом не повреждает ДНК бактерии, из которой она была выделена.
В дальнейшем было выделено несколько сотен рестриктаз из разных бактерий, способных узнавать и расщеплять специфические последовательности нуклеотидов (сайты узнавания) в молекуле ДНК. Некоторые из этих ферментов узнают группы из четырех нуклеотидов, другие - из шести и даже более нуклеотидов. Естественно, рестриктазы, распознающие группы из четырех нуклеотидов, вносят в молекулу ДНК, выделенную из какого-либо организма, больше разрывов, чем те, которые узнают шесть и более нуклеотидов.
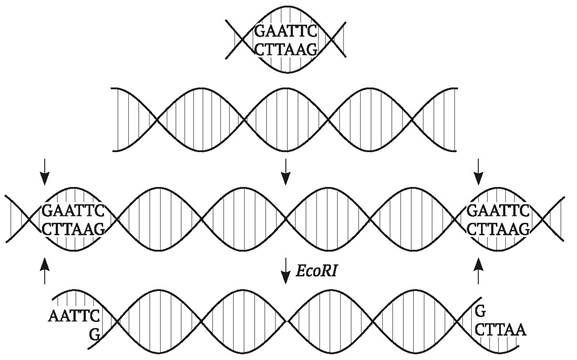
Для целей генной инженерии наибольшее значение имело выделение рестриктаз, которые дают фрагменты с так называемыми «липкими» (комплементарными) концами. В случае их использования разрывы ДНК происходят в местах, расположенных наискось: на концах каждого из полученных фрагментов остаются короткие одноцепочечные хвосты из нескольких нуклеотидов (табл. 9.1). Если объединить в одной пробирке фрагменты ДНК любого происхождения, полученные с помощью одной и той же рестриктазы, дающей «липкие» концы, то эти фрагменты соединятся между собой. Однако водородные связи между комплементарными азотистыми основаниями недостаточно прочны, чтобы стабильно удерживать два объединенных фрагмента ДНК. Для воссоединения фосфодиэфирных связей между концами сшиваемых фрагментов ДНК необходим фермент ДНК-лигаза. Этот фермент был впервые выделен в 1967 г. из культуры E. coli, инфицированной бактериофагом Т4, при изучении энзимологии процесса репликации ДНК (т. е. еще до того, как научились разрезать ДНК).
Таблица 9.1. Нуклеотидные последовательности, распознаваемые некоторыми ферментами рестрикции, и характер получаемых концов ДНК (Б. Глик, Дж. Пастернак, 2002)
Рестриктаза |
Сайт узнавания |
Характер получаемых концов ДНК |
EcoRI |
Г↓А-А-Т-Т-Ц Ц-Т-Т-А-А↑Г |
«Липкие» концы с 5'-фосфатной группой |
BamHI |
Г↓Г-А-Т-Ц-Ц Ц-Ц-Т-А-Г↑Г |
«Липкие» концы с 5'-фосфатной группой |
PstI |
Ц-Т-Г-Ц-А↓Г Г↑А-Ц-Г-Т-Ц |
«Липкие» концы с 3'-фосфатной группой |
Sau3AI |
↓Г-А-Т-Ц Ц-Т-А-Г ↑ |
«Липкие» концы с 5'-фосфатной группой |
Notl |
Г↓Ц-Г-Г-Ц-Ц-Г-Ц Ц-Г-Ц-Ц-Г-Г-Ц↑Г |
«Липкие» концы с 5'-фосфатной группой |
РvuII |
Ц-А-Г↓Ц-Т-Г Г-Т-Ц↑Г-А-Ц |
«Тупые» концы |
НраI |
Г-Т-Т↓-А-Ц Ц-А-А↑Т-Т-Г |
«Тупые» концы |
НаеIII |
Г-Г↓Ц-Ц Ц-Ц↑Г-Г |
«Тупые» концы |
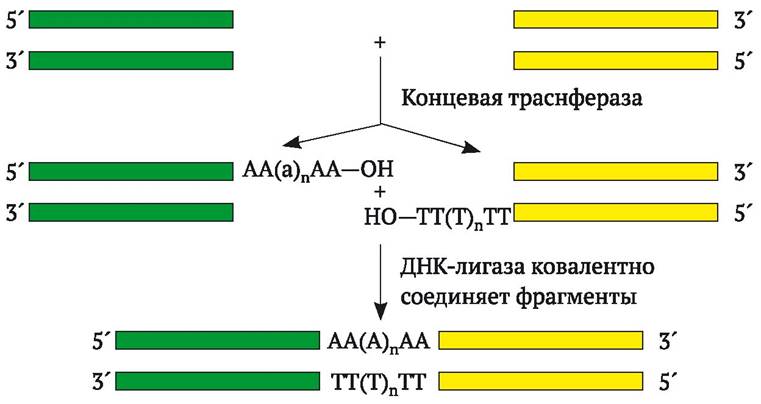
Для генной инженерии большое значение имеет использование еще одного фермента - концевой трансферазы из тимуса теленка, который катализирует присоединение нуклеотидов к 3'-концам цепей ДНК. Благодаря этому из «тупого» конца ДНК несложно сделать «липкий». Если к обоим 3'-концам двухцепочечного фрагмента присоединить несколько расположенных друг за другом адениновых нуклеотидов (поли-А), а к другому фрагменту ДНК присоединить - поли-Т, то при смешивании этих двух фрагментов они соединятся между собой, поскольку их концы комплементарны друг другу (рис. 9.1).
Рис. 9.1. Легирование двух концов ДНК с «тупыми» концами путем присоединения к ним фрагментов роlу-dА или роlу-dТ (по Дж. Уотсону и др., 1986)
В генно-инженерных работах часто используют еще один метод произвольного сшивания фрагментов ДНК, основанный на применении так называемых линкеров - искусственных сайтов рестрикции. Линкеры представляют собой короткие фрагменты ДНК (олигонуклеотиды), состоящие из 8-10 нуклеотидных пар. Их синтезируют искусственно. При этом состав и последовательность нуклеотидов в них соответствует сайту рестрикции одной из рестриктаз, создающей «липкие» концы и используемой при выделении фрагмента ДНК, который предполагается пришить к какому-то определенному фрагменту ДНК. Линкеры пришивают к этому последнему фрагменту ДНК с помощью концевой трансферазы и ДНК-лигазы, а затем расщепляют их рестриктазой. В результате этот фрагмент приобретает «липкий» конец нужной нам конфигурации (рис. 9.2). Линкеры, имеющие несколько сайтов узнавания рестриктазами, называются полилинкерами.
Рис. 9.2. Линкеры: олигонуклеотиды, содержащие сайт рестрикции, добавляемые с помощью концевой трансферазы и ДНК-лигазы к «тупым» концам фрагмента ДНК (после обработки соответствующей рестриктазой фрагмент ДНК будет иметь «липкие» концы, совместимые с концами ДНК, разрезанной той же самой рестриктазой)
Одна какая-либо рестриктаза вносит строго определенное количество разрывов в определенных местах молекулы ДНК какого-либо анализируемого организма. В этом можно убедиться, если образовавшиеся после обработки ферментом фрагменты ДНК разделить с помощью гель-электрофореза.
Принцип этого распространенного аналитического метода, который используют для исследования белков и нуклеиновых кислот, заключается в следующем. Компоненты смеси разделяют путем пропускания ее через гель - полужидкую среду с сетчатой пространственной структурой, которую получают путем полимеризации агара, агарозы, сополимеризации акриламида и бисакриламида, и др. Обычно гели готовят в виде тонких пластинок, имеющих у одного края лунки для нанесения препарата. Находящиеся в буферном растворе макромолекулы обладают электрическим зарядом. Когда через гель с нанесенным раствором пропускают электрический ток, то компоненты смеси начинают перемещаться в электрическом поле. Фрагменты ДНК или отдельные протеины, содержащиеся в анализируемой смеси, имеют разную длину, а, следовательно, и размеры. Скорость миграции компонентов смеси в геле пропорциональна их размерам: более мелкие фрагменты будут перемещаться быстрее. В результате по окончании электрофореза, когда наиболее быстрые компоненты смеси достигнут противоположного края гелевой пластинки, остальные равномерно распределятся по ее длине в соответствии со своими размерами. После их окрашивания специальными красителями можно наблюдать на геле спектр четко различимых полос, расположенных одна за другой, каждая из которых соответствует отдельному компоненту смеси.
Электрофоретические спектры рестрикционных фрагментов для какой- либо рестриктазы и для ДНК какого-либо организма строго специфичны. Сколько бы раз ни получали препараты ДНК, сколько бы раз ни обрабатывали ее рестриктазой, в любом случае при электрофорезе мы получим один и тот же спектр, состоящий из определенного количества специфических полос. На основании этого правила строят так называемые рестрикционные карты молекул ДНК, на которые наносят в установленной последовательности сайты рестрикции различных рестриктаз. Такие карты используют в молекулярной генетике для характеристики отдельных ДНК и в генной инженерии для подбора эндонуклеаз при выделении отдельных генов.
Исключительно важной особенностью электрофоретического разделения рестрикционных фрагментов ДНК является то, что при электрофорезе эти фрагменты не разрушаются и их можно выделить (элюировать) из геля в виде биологически активных двухцепочечных молекул. Фрагменты ДНК, выделенные из одной какой-либо полосы, будут строго идентичны друг другу. Их далее можно анализировать на предмет определения последовательности нуклеотидов, использовать в качестве материала для построения генетических конструкций и т. д.
Объединение разных молекул ДНК с помощью методов, описанных выше, само по себе бесполезно, если полученные рекомбинантные ДНК не внести в живую клетку, не заставить их там реплицироваться, и, что самое важное, заставить привнесенные гены работать: синтезировать мРНК, транслировать ее в рибосомах с образованием протеина - продукта трансгена.
Первая функционально активная молекула рекомбинантной ДНК была получена в 1973 г. Г. Бойер и С. Коэн (США) сумели соединить фрагменты ДНК плазмид разных бактерий, полученных путем их разрезания рестриктазой, дающей «липкие» концы, и ввели ее в Е. соli. Они обнаружили, что такая химерная плазмида могла успешно функционировать, реплицироваться и передаваться потомству как естественным путем, так и с помощью человека (S. Cohen и др., 1973). Это означало, что таким образом можно получать многочисленные копии любых генов, т. е. клонировать гены, нарабатывать достаточные объемы генетического материала для последующего изучения и возможных манипуляций с ними.
Выбор плазмиды американскими учеными в качестве вектора клонирования оказался весьма удачным. Именно плазмидные векторы в настоящее время используют в генетической инженерии чаще всего. То же относится к бактерии Е. соli, которую чаще всего используют в качестве «места» клонирования. Попытаемся разобраться, что лежит в основе их успеха.
Для того чтобы рекомбинантная ДНК имела возможность реплицироваться в живой клетке, она должна содержать ту часть естественной молекулы ДНК, которая ответственна за начало, инициацию этого процесса. Вероятность того, что она окажется в случайных рестрикционных фрагментах ДНК, ничтожна. В то же время сайт начала репликации имеется в любой бактериальной плазмиде (его обозначают как оri). Поэтому, если в плазмиду встроить нужный фрагмент ДНК, она обеспечит репликацию как своей собственной ДНК, так и встроенного фрагмента.
Плазмиды - это внехромосомные автономно реплицирующиеся двухцепочечные кольцевые молекулы ДНК. Они присутствуют в клетках практически всех бактерий. Размеры плазмид от менее 1 до более 500 тыс. пар нуклеотидов (т. п. н.). Они могут быть представлены в клетке одной, несколькими, или многими (10-100) копиями. На их долю приходится до 5 % суммарной клеточной ДНК. Малокопийные плазмиды реплицируются с той же скоростью, что и бактериальная хромосома, многокопийные плазмиды способны реплицироваться независимо.
Функции плазмид весьма разнообразны. Есть плазмиды так называемые F-факторы (F-плазмиды), которые отвечают за прохождение полового процесса у бактерий (конъюгацию), при котором осуществляется перенос плазмид из одной бактериальной клетки в другую, а также части генетической информации, находящейся на бактериальной хромосоме. Имеются плазмиды, которые определяют устойчивость бактерий к антибиотикам (R-плазмиды) или их способность утилизировать, перерабатывать необычные вещества, например, нефтепродукты, пестициды и другие загрязнители окружающей среды (плазмиды деградации). Расположение таких генов именно на плазмидах, а не на хромосоме неслучайно. Высокая копийность плазмид обеспечивает клетке возможность синтеза большого количества ферментов, способных нейтрализовать антибиотики или расщеплять пестициды.
Некоторые плазмиды, называемые эписомами, могут обратимо включаться в бактериальную хромосому (к ним относятся упомянутые выше F-факторы). Если две плазмиды не могут сосуществовать в одной клетке, то их относят к одной группе несовместимости. Плазмиды из разных групп несовместимости могут находиться в одной клетке независимо от числа копий. Отмечены случаи, когда у бактерий обнаруживали до 8-10 разных групп плазмид, выполняющих свои функции, и представленных характерным для каждой группы числом копий.
Плазмиды могут передаваться от одной бактериальной клетки к другой, в том числе и неродственной. Чтобы интенсифицировать процесс внедрения плазмид в бактериальные клетки, в экспериментах используют специальные приемы. Например, подвергают клетки высокотемпературному воздействию и обрабатывают их хлористым кальцием (СаСl2).
У одних плазмид сайт инициации репликации (оri) строго специфичен, плазмиды с таким сайтом способны реплицироваться только в клетках одного вида бактерий. У других плазмид этот сайт менее специфичен, и они способны реплицироваться в самых разных бактериальных клетках. Соответственно различают плазмиды с узким и широким спектром хозяев. Последняя группа плазмид лежит в основе быстрой передачи генов устойчивости к антибиотикам к ранее чувствительным к ним болезнетворным микроорганизмам.
В качестве векторов клонирования используют специально отобранные и генетически модифицированные плазмиды. Основные требования к вектору следующие: 1) небольшой размер, поскольку эффективность клонирования чужеродной ДНК в Е. coli значительно снижается при общей длине привнесенной рекомбинантной плазмиды более 15 т. п. н.; 2) наличие уникального сайта рестрикции, в который может быть осуществлена вставка; 3) наличие одного или нескольких селективных генетических маркеров для идентификации трансформированных клеток (т. е. включивших привнесенные гены). В качестве последних чаще всего используют гены устойчивости к антибиотикам или гербицидам.
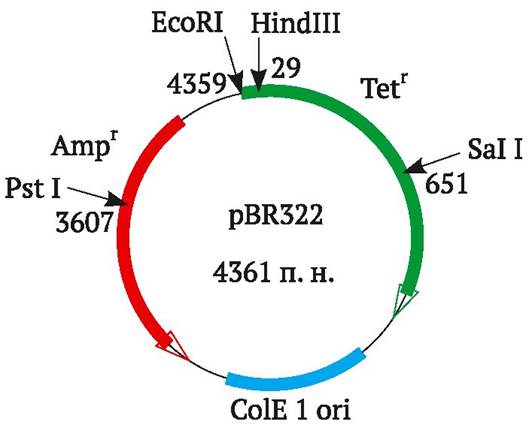
Процедура клонирования фрагментов ДНК осуществляется следующим образом. Первый этап включает разрезание кольцевой молекулы ДНК плазмиды, которую используют в качестве вектора клонирования. Плазмида, как правило, содержит два маркерных гена, например, гены устойчивости к двум разным антибиотикам (рис. 9.3). Обычно место расщепления плазмидной ДНК находится в пределах структурной последовательности одного из ее маркерных генов. Такой же самой рестриктазой, образующей «липкие» концы, разрезают ДНК, фрагменты которой предполагается клонировать (донорной ДНК), и смешивают с разрезанной плазмидной ДНК. Поскольку «липкие» концы этих двух ДНК взаимно комплементарны, они соединяются с образованием гибридных молекул. Процесс сшивки закрепляют путем добавления в раствор фермента ДНК-лигазы. В результате образуется смесь разных комбинаций плазмидной и донорной ДНК, а также такие нежелательные продукты, как слившиеся фрагменты только донорной ДНК или восстановленные кольцевые молекулы плазмидной ДНК.
Рис. 9.3. Основные компоненты плазмиды pBR322, чаще всего используемой в качестве вектора клонирования генов в E. coli (плазмида включает ori-сайт и гены устойчивости к антибиотикам тетрациклину (Tetr) и ампициллину (Ampr), она содержит уникальные сайты рестрикции для более чем 40 рестриктаз, часть из которых расположена внутри генов устойчивости к антибиотикам, на рисунке стрелками отмечены некоторые из них) (http:// faculty.tru.ca/dnelson/courses/biol335)
Рекомбинантные плазмиды, содержащие встроенные фрагменты донорной ДНК, далее вводят в клетку-хозяина, обычно это - Е. coli. Для этого бактерии смешивают с раствором плазмид, в который добавляют хлористый кальций, подвергают клетки нагреванию. Тем не менее эффективность трансформации (внедрения чужеродной ДНК в клетку) невысокая: порядка одной клетки на тысячу. Что получается в результате? Большинство бактериальных клеток остаются интактными. Небольшое количество клеток содержит различные рекомбинантные плазмиды. Могут также быть клетки, содержащие нежелательные продукты: интактные векторные плазмиды, фрагменты донорной ДНК. Судьба последних очевидна. Чужеродная ДНК, не имеющая сайта ori, не может реплицироваться в бактериальной клетке и разрушается под действием эндонуклеаз.
Таким образом, задача сводится к тому, чтобы разделить первые три типа бактериальных клеток. Для этого их последовательно высевают на питательные среды, содержащие антибиотики, являющиеся селективными агентами в нашем эксперименте. Нетрансформированные клетки чувствительны к обоим антибиотикам, клетки с интактными плазмидами, напротив, устойчивы к обоим антибиотикам, а клетки с рекомбинантными плазмидами устойчивы только к тому антибиотику, ген устойчивости к которому не поврежден в результате рестрикции и вставки фрагмента донорной ДНК. Поскольку рекомбинантная плазмида содержит все необходимое для инициации репликации, то она может реплицироваться в бактериальных клетках, т. е. осуществлять клонирование своей, а также встроенной в нее донорной ДНК.
Помимо бактериальных плазмид для целей клонирования генов используют и другие векторные системы: фаги (в них можно клонировать более длинные, нежели в плазмидах, фрагменты донорной ДНК: до 20 т. п. н.), плазмиды-космиды, содержащие cos-участок генома фага, участвующий в упаковке ДНК в фаговую головку (в них можно клонировать фрагменты ДНК до 40 т. п. н.), а также так называемые искусственные хромосомы на основе фага Р1 (можно клонировать фрагменты ДНК длиной 100-300 т. п. н.), на основе F-плазмиды Е. coli (BAC - бактериальная искусственная хромосома, емкость 150-300 т. п. н.), дрожжей (YAC-дрожжевая искусственная хромосома, в ней можно клонировать фрагменты эукариотической ДНК длиной порядка 800 т. п. н. и более). В качестве клеток-хозяев для клонирования генов помимо Е. coli используют также Bacillus subtilis, дрожжи Saccharomyces cerevisiae и др.
9.2. Выделение генов
С помощью набора разных рестриктаз получают многие тысячи фрагментов ДНК, содержащих самые разнообразные гены. Каким образом в этой смеси найти и выделить фрагмент, который кодирует нужный ген, а также регуляторные последовательности, необходимые для его функционирования в клетке? Это один из наиболее сложных и дорогостоящих этапов генетической инженерии. Для решения проблемы идентификации генов разработаны и успешно применяются многие методы, без которых эта задача оставалась бы практически неразрешимой. Так, ученые научились определять последовательность аминокислот в белковых молекулах и последовательность нуклеотидов в нуклеиновых кислотах ДНК и РНК. Разработаны методы получения молекул ДНК (кДНК), комплементарных определенным молекулам мРНК (метод обратной транскрипции). Более того, в настоящее время вполне рутинной процедурой стал химический синтез отдельных генов. Большинство названных методов, а также радиоавтография, хемилюминесцентное маркирование, различные варианты гель-электрофореза, полимеразная цепная реакция и другие в полной мере используются для выделения и идентификации отдельных генов. Рассмотрим подробнее, каким образом все это используется на практике.
Возможны два основных подхода в выделении гена: либо его синтезируют, либо выбирают среди рестрикционных фрагментов геномной ДНК тот из них, который содержит нужный ген. Чаще всего эти подходы используют в различных сочетаниях с учетом конкретной ситуации. Есть гены и протеины - продукты генов, которые изучены очень хорошо, а есть такие, про которые практически ничего не известно. Прокариотические гены выделять в общем проще, чем эукариотические, особенно в тех случаях, когда они расположены на плазмидах: поле поиска намного уже. Большое значение при выделении генов имеет степень и характер их экспрессии: проще выделять те из них, что активно транскрибируются в каких-то определенных тканях, благодаря чему в клетках этих тканей преобладает мРНК искомого гена. В любом случае выделение генов - сложный творческий процесс, требующий от исследователя глубоких знаний и владения широким арсеналом современных методов.
9.2.1. Искусственный синтез генов
Чтобы синтезировать ген искусственно, необходимо знать его последовательность нуклеотидов. В 1977 г. Ф. Сэнгер с сотрудниками (F. Sanger и др., 1977) предложили относительно простой, недорогой и надежный метод определения последовательности нуклеотидов ДНК (секвенирование ДНК) с помощью терминирующих дидезоксинуклеотидов (метод ферментативного копирования с остановкой удлинения цепи, осуществляемого ДНК- полимеразой). За его разработку он был удостоен Нобелевской премии по химии за 1980 г. (совместно с W. Gilbert). Возможность прямого секвенирования ДНК произвела настоящую революцию в молекулярной генетике. С помощью метода Сэнгера (и его последующих модификаций) определена последовательность нуклеотидов не только отдельных генов, но и целых геномов, в том числе генома человека, а также ряда видов животных и растений. Вся эта бесценная информация хранится в международных базах данных и доступна в Интернете для любого исследователя. Информация о нуклеотидной последовательности фрагмента ДНК дает возможность идентифицировать кодирующую область гена, его регуляторные элементы, подобрать потенциальные праймеры для полимеразной цепной реакции (ПЦР), выявить мутационные изменения в гене, определить последовательность аминокислот в протеине-продукте гена.
Белки-продукты многих генов хорошо известны и изучены, в том числе на предмет последовательности аминокислот в их молекулах. Зная эту последовательность и имея таблицу генетического кода, можно определить последовательность нуклеотидов в мРНК и в ДНК, которая должна при трансляции обеспечить синтез белковой молекулы именно с такой последовательностью аминокислот. Безусловно, искусственный ген не будет идентичен натуральному по той простой причине, что генетический код вырожденный: одной аминокислоте может соответствовать несколько сочетаний из трех нуклеотидов. Следует также иметь в виду, что гены высших организмов содержат некодирующие участки - интроны. Тем не менее важен результат: искусственный ген, если его заставить работать, будет образовывать мРНК, с которой будет при трансляции синтезирован протеин, идентичный натуральному.
Современные ДНК-синтезаторы способны присоединять один нуклеотид к цепочке ДНК в течение нескольких минут, формируя фрагменты ДНК длиной более ста пар нуклеотидов. Соединяя отдельные фрагменты ДНК, можно получить полноразмерный ген. Таким методом был синтезирован, в частности, хорошо известный ген проинсулина человека, а также гены ряда других протеинов - продуктов современной биотехнологии.
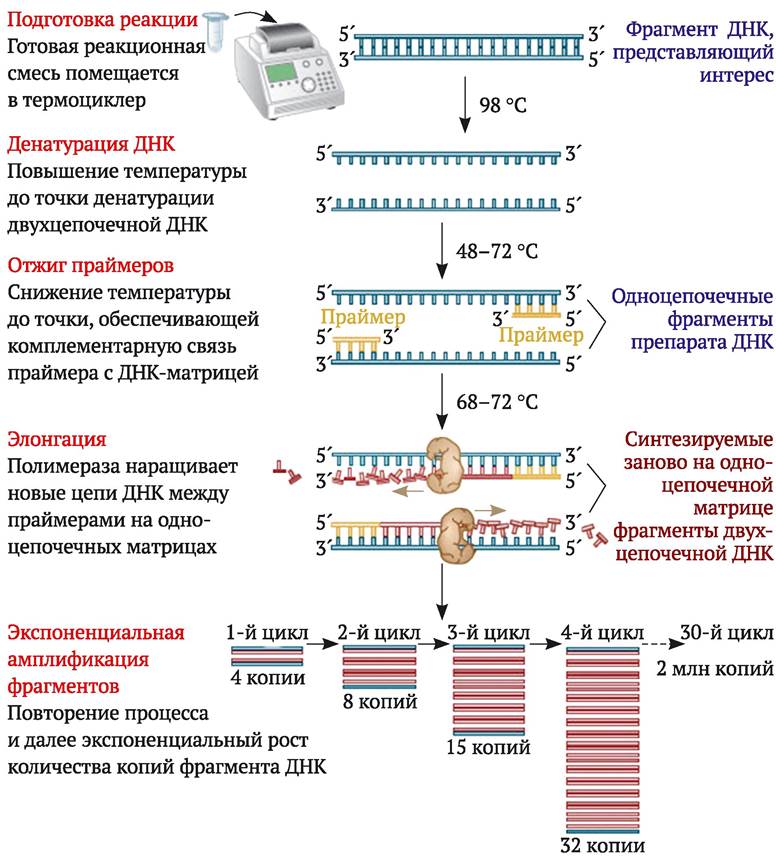
Второй способ искусственного синтеза гена основан на использовании метода полимеразной цепной реакции, предложенного K. B. Mullis и другими (1986). Метод ПЦР позволяет получать in vitro большое количество копий определенных последовательностей ДНК. Амплификация ДНК происходит в смеси, содержащей образец ДНК, два синтетических олигонуклеотидных праймера (длиной около 20 нуклеотидов), комплементарных участкам ДНК из противоположных цепей, фланкирующих (находящихся на концах) фрагмент, который предполагается амплифицировать, термостабильную ДНК- полимеразу (Таq-полимеразу), дезоксирибозидтринуклеотидфосфаты (dATP, dGTP, dCTP, dTTP), ионы магния и буферный раствор.
Первый этап ПЦР состоит в тепловой денатурации образца ДНК путем нагревания его до температуры около 95 °С. В результате температурного воздействия образуется однонитчатая ДНК. Второй этап - ренатурация (отжиг). Температуру смеси понижают до температуры, специфичной для ренатурации конкретно используемой пары фланкирующих последовательностей ДНК (обычно между 50 и 64 °С). При этом праймеры соединяются (гибридизуются) с комплементарными последовательностями образца ДНК. Третий этап - синтез (элонгация ДНК). Температуру повышают до 72-75 °С, которая является оптимальной для термостабильной ДНК-полимеразы. На этом этапе происходит инициированное праймерами достраивание ДНК (синтез комплементарной цепи ДНК). Затем весь цикл многократно повторяют (рис. 9.4). Все реакции проводят в специальных пробирках в автоматическом термоциклере (амплификаторе), который обеспечивает точное соблюдение режима амплификации. Режим амплификации (температура и продолжительность отдельных этапов амплификации, количество циклов), а также состав реакционной смеси оптимизируют для каждого конкретного случая. В результате многократного повторения реакций амплификации ДНК (обычно выполняют около 40 циклов) образуются многочисленные копии последовательности ДНК, заключенной между праймерами. Ее несложно идентифицировать в виде отдельной полосы на электрофореграмме, определить с помощью специальной калибровки ее длину (количество пар нуклеотидов). Можно элюировать ее из геля и использовать для изучения (например, секвенирования) или генетических манипуляций.
Рис. 9.4. Схема амплификации ДНК методом полимеразной цепной реакции
Метод полимеразной цепной реакции ДНК нашел исключительно широкое применение в молекулярно-генетических исследованиях. Он лежит в основе многочисленных диагностических методов, позволяющих выявлять в анализируемой ДНК определенные гены, ДНК различных патогенов. За разработку этого метода К. B. Mullis был удостоен Нобелевской премии по химии за 1993 г.
Если в международных базах данных содержится информация о сиквенсе интересующего нас гена какого-либо организма (или близких в систематическом отношении организмов), то можно с помощью специальных компьютерных программ подобрать праймеры, необходимые для амплификации кодирующей последовательности ДНК, используя в качестве исходной матрицы ДНК этого организма. Следует, правда, иметь в виду, что с помощью описанной технологии можно получить относительно короткие фрагменты ДНК (не более 3000 пар нуклеотидов). В связи с этим рекомендуют амплифицировать несколько перекрывающихся фрагментов ДНК кодирующей последовательности гена, а затем получить с помощью технологии рекомбинатных ДНК полноразмерный ген.
В реакции амплификации в качестве матрицы может быть использована его мРНК. В отдельных случаях мРНК некоторых генов можно выделить непосредственно из клеток, в которых они могут быть в большом количестве. Соответствующие мРНК выделяют, тщательно очищают и используют для синтеза комплементарной ДНК с помощью фермента обратной транскриптазы, или ревертазы. Этот фермент был выделен в 1970 г. из РНК-содержащих онкогенных вирусов (его первооткрыватели Н. М. Теmin и D. Ваltimor были удостоены Нобелевской премии в области физиологии и медицины за 1975 г.).
Для старта полимеразной цепной реакции ферменту нужна «затравка» (праймер) в виде небольшого отрезка двухцепочечной молекулы (рис. 9.5).
Рис. 9.5. Получение кДНК с РНК-матрицы с помощью фермента обратной транскриптазы: а - к эукариотической мРНК, содержащей поли-А хвост, добавляют праймер поли-Т (Роlу-dТ), комплементарный поли-А; б - праймер соединяется с поли-А; в - в присутствии фермента обратной транскриптазы происходит синтез молекулы ДНК, комплементарной мРНК; г - РНК удаляют с помощью раствора щелочи; д - шпилька служит праймером для синтеза комплементарной нити ДНК в присутствии фермента ДНК полимеразы I; е - шпильку удаляют с помощью фермента Sl-нуклеазы: получают кДНК (по Дж. Уотсону и др., 1986)
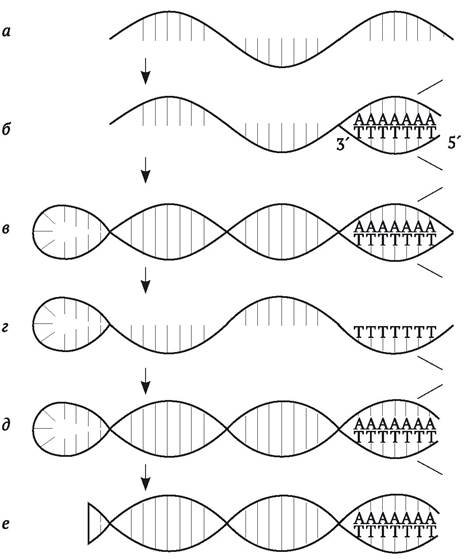
Эукариотические мРНК содержат, как правило, на своем 3'-конце так называемый поли-А хвост - последовательность, состоящую из остатков аденина. Если к мРНК добавить искусственно синтезированные короткие олигонуклеотиды, состоящие из остатков тимина (поли-Т), они будут в силу комплементарности соединяться с поли-А, образуя двойную цепочку, которая необходима для инициации ферментативной реакции полимеризации ДНК нуклеотидов. В результате образуется гибридная двухцепочечная РНК-ДНК молекула. Далее мРНК отщепляют от этой молекулы с помощью NaOH (нить ДНК при этом не повреждается). После этого остается одноцепочечная молекула ДНК, на конце которой имеется так называемая «шпилька» (короткий отрезок двухцепочечной ДНК), служащая «затравкой» для синтеза второй цепи ДНК под действием фермента ДНК-полимеразы I. «Шпильку» далее удаляют с помощью фермента S1-нуклеазы, которая разрушает одноцепочечные ДНК. В конце концов, получается двухцепочечная молекула ДНК, одна из нитей которой комплементарна исходной мРНК. Такую ДНК называют кДНК (комплементарная ДНК) и она, в общем, соответствует структурному гену, с которого транскрибировалась мРНК. Разумеется, она не будет содержать интроны, которые могли быть у натурального гена. Описанный метод синтеза кДНК получил название ОТ-ПЦР - ПЦР с обратной транскрипцией (от англ. RT-PCR - reverse transcription PCR).
Полученную кДНК с помощью технологии рекомбинантных ДНК можно соединить с соответствующими регуляторными элементами, встроить в векторную плазмиду и перенести в клетки бактерий, где они будут реплицироваться и экспрессироваться. По их продукту можно будет окончательно убедиться, ту ли, нужную мРНК, выделили.
9.2.2. Выделение генов из смеси рестрикционных фрагментов геномной ДНК
Для выделения нужного гена из смеси рестрикционных фрагментов используют меченые зонды - фрагменты ДНК, комплементарные искомым генам (целиком или отдельным их частям). Зонды метят с помощью радиоактивного фосфора или хемилюминесцентных меток. В основе метода лежит свойство одноцепочечных молекул ДНК соединяться (гибридизироваться) в случае их комплементарности.
Для перевода молекулы ДНК в одноцепочечную форму ее подвергают тепловому воздействию или обработке щелочью. В этих условиях водородные связи между основаниями разрываются и цепи расходятся (процесс денатурации ДНК). Если теперь медленно снизить температуру, то произойдет их восстановление (ренатурация). Однако если при этом в растворе присутствует одноцепочечный ДНК-зонд, он тоже будет ренатурировать с ДНК-матрицей, специфически связываясь с комплементарными участками.
Нужный ген может быть выделен или до, или после процедуры клонирования в E. coli смеси рестрикционных фрагментов геномной ДНК. В первом случае фрагменты ДНК, полученные после обработки рестриктазами, разделяют в соответствии с их размерами с помощью электрофореза в агарозном геле. Поскольку проводить описанные выше процедуры гибридизации ДНК с зондами в геле невозможно, ДНК после электрофореза переносят на более подходящие для этих целей носители: нитроцеллюлозную или нейлоновую мембраны. Фрагменты ДНК прочно связываются с ними и не могут изменять свое положение, например, из-за диффузии. Процедуру переноса фрагментов ДНК с агарозного геля на нитроцеллюлозные мембраны и проведения гибридизации с зондами разработал Э. Саузерн (E. Sauthern, 1975). Благодаря простоте и высокой эффективности этот метод нашел широкое применение в генно-инженерных исследованиях. Его название - Саузерн-блоттинг - стало нарицательным. Два других аналогичных метода, в которых используются РНК и белки, получили названия соответственно нозери-блоттинг (северный блоттинг) и вестерн-блоттинг (западный блоттинг), что является игрой слов (Саузерн (southern) - южный).
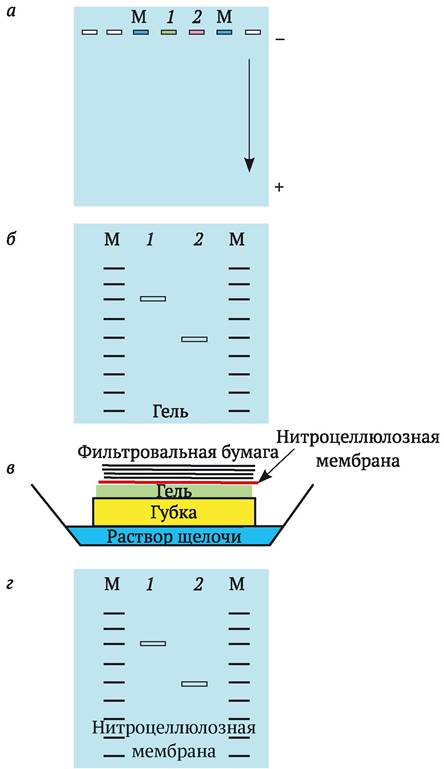
Саузерн-блоттинг проводят следующим образом (рис. 9.6). Двунитчатую ДНК на агарозном геле денатурируют с помощью NaOH, чтобы образовались однонитчатые молекулы. Затем агарозный гель помещают на влажную губку, расположенную в кювете с раствором щелочи, сверху кладут нитроцеллюлозный фильтр, далее несколько слоев сухой фильтровальной бумаги и весь этот сэндвич прижимают грузом. Сухая фильтровальная бумага начинает втягивать влагу из расположенных ниже слоев. Создается поток щелочи снизувверх из губки через гель, нитроцеллюлозную мембрану к фильтровальной бумаге. Этим потоком фрагменты ДНК вымываются из геля и закрепляются на нитроцеллюлозе именно в той последовательности, как они расположились после электрофореза. Однонитчатые фрагменты ДНК фиксируют на нитроцеллюлозной мембране с помощью нагревания. На ней и проводят гибридизацию с меченым ДНК-зондом.
Рис. 9.6. Блоттинг ДНК по Саузерну: а - нанесение меченых фрагментов ДНК на агарозный гель (М - маркер молекулярного веса с меткой; 1 и 2 - пробы изучаемых фрагментов ДНК, стрелкой показано направление перемещения ДНК в геле от анода к катоду); б - результат электрофореза ДНК в агарозном геле; в - размещение геля в кювете для блот-гибридизации (стрелкой показано движение раствора щелочи через гель и перенос фрагментов ДНК на нитроцеллюлозную мембрану); 2 - фрагменты ДНК и молекулярных маркеров после переноса на мембрану
В случае если зонд находит комплементарный ему фрагмент ДНК, происходит их связывание. Местоположение связавшегося зонда определяют, приложив к нитроцеллюлозной мембране рентгеновскую пленку, в которой под действием радиоактивного излучения из бромида серебра образуется металлическое серебро. Засвеченные участки, соответствующие радиоактивным зонам, наблюдаются визуально после проявления пленки. При использовании хемилюминисцентных меток их местоположение определяют в ультрафиолетовом или лазерном свете.
Для того чтобы выделить нужный фрагмент ДНК, повторно проводят электрофорез рестрикционных фрагментов ДНК или берут второй гель, если электрофорез проводился одновременно в двух гелях. Этот гель, который не подвергался ДНК-ДНК гибридизации, имеет интересующий сегмент в том же месте, что и в проанализированном с помощью ДНК-зонда геле. Сегмент вырезают из геля и извлекают из него ДНК, которую можно далее клонировать, как это было описано выше.
9.2.3. Выделение генов из клонотеки
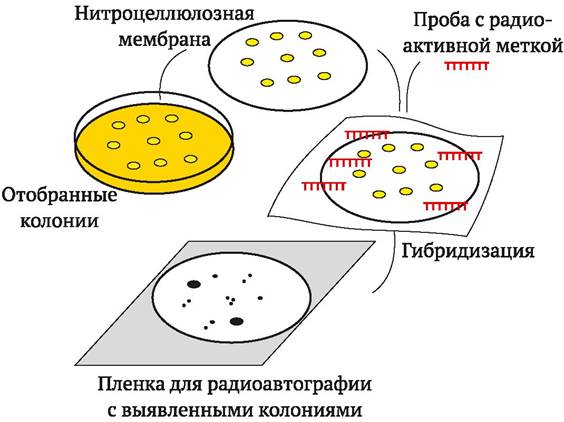
ДНК-ДНК гибридизацию применяют и для выделения искомых фрагментов ДНК после того, как они были клонированы. Этот подход получил название «дробовик» (shortgun) и его суть состоит в создании и изучении так называемой геномной библиотеки, или клонотеки генов. Клонотека представляет собой колонии бактерий, клетки которых содержат рекомбинантные плазмиды со встроенными в них разными рестрикционными фрагментами ДНК организма. Для выделения генов из клонотеки электрофорез и Саузерн-блоттинг не требуются, поскольку фрагменты ДНК разделены пространственно по разным колониям бактерий-хозяев. Эти колонии переносят непосредственно на нитроцеллюлозный фильтр, размещая их в определенном порядке. При подготовке проб к анализу проводят лизис бактериальных клеток и денатурацию содержащейся в них ДНК. Затем осуществляют гибридизацию с меченым зондом и определяют таким образом, в какой колонии клонирован интересующий фрагмент ДНК. Этот метод получил название дот-блоттинга (от англ. dot - точка, пятнышко) (рис. 9.7).
Рис. 9.7. Дот-блоттинг (пояснения в тексте)
Как при выделении генов из клонотек, так и из смеси разделенных с помощью электрофореза рестрикционных фрагментов ДНК, следует иметь в виду, что положительный гибридизационный сигнал может быть получен для нескольких клонов (или полос электрофоретического спектра). Объяснить это несложно: рестриктаза могла внести несколько разрезов по длине гена, или в результате частичного гидролиза образовались фрагменты ДНК, содержащие искомый ген, разной длины. В связи с этим необходимо определить, какой именно клон или фрагмент ДНК содержит ген целиком.
С помощью гель-электрофореза и секвенирования каждого выделившегося фрагмента ДНК (вставки) идентифицируют аналогичные фрагменты или фрагменты с перекрывающимися последовательностями. Проводят сшивку отдельных коротких фрагментов и клонирование полученных последовательностей с тем, чтобы составить полный ген. Если вставка анализируемой ДНК в каком-либо из клонов достаточно велика и вполне может содержать весь ген, проводят ее секвенирование, чтобы найти старт- и стоп-кодоны и убедиться в наличии полноразмерной нуклеотидной последовательности, кодирующей искомый белок. Результаты этого анализа позволяют также наметить план дальнейшей модификации выделенного фрагмента ДНК. Ведь для целей генетической инженерии важно иметь фрагмент ДНК, не содержащий последовательностей, которые не имеют отношения к нужному нам гену.
При создании зондов наибольшую трудность представляет подбор нужного фрагмента ДНК или РНК. Наиболее простой метод состоит в использовании ранее клонированных и идентифицированных генов, можно даже близкородственных видов (гетерологичный зонд). В последнем случае условия гибридизации нужно подбирать таким образом, чтобы она могла происходить при существенном расхождении в последовательности искомой ДНК и зонда.
Зонд можно получить с помощью химического синтеза, основываясь на известной аминокислотной последовательности протеина-продукта искомого гена. Выбирают небольшую часть белковой молекулы длиной 5-6 аминокислот. В соответствии с последовательностью этих аминокислот определяют все возможные последовательности нуклеотидов в том участке мРНК, который кодирует данную область белковой молекулы. Затем из радиоактивно меченых нуклеотидов синтезируют соответствующие олигонуклеотиды длиной не менее 16 звеньев, один из которых будет полностью комплементарен участку искомого гена.
Для зонда можно также использовать препараты мРНК или кДНК, полученные, как это было описано выше. Получение большого количества разнообразных кДНК, создание клонотек кДНК, их изучение имеет в генетической инженерии большое значение. По сравнению с клонотеками рестрикционных фрагментов, большинство из которых может и не содержать отдельных генов или содержать только их части, библиотеки кДНК имеют несомненное преимущество. В случае с кДНК речь идет о последовательностях, кодирующих, образно говоря, гены в чистом виде, без интронов, и не относящихся к гену последовательностей ДНК.
Главная задача - идентифицировать эти гены. Наиболее надежный способ решения задачи - добиться экспрессии (т. е. транскрипции и образования протеина-продукта гена) клонированной кДНК в клетке-хозяине. Для этих целей используют специальные клонирующие векторные плазмиды (экспрессирующие вектора), у которых место встраивания клонируемой ДНК расположено непосредственно за промотором, захватывая лишь несколько нуклеотидов прокариотического гена. Как, указывалось выше, бактерии в природе не могут экспрессировать эукариотические гены. Но в случае с данными плазмидами эукариотический ген становится как бы частью прокариотического гена со своим промотором. В результате активности такого гибридного гена в бактерии образуется рекомбинантный эукариотический белок, имеющий лишь несколько аминокислот, кодируемых прокариотическим геном.
Для идентификации эукариотического протеина используют метод, аналогичный дот-блоттингу, в котором в качестве зонда применяют меченые антитела определенных известных белков (метод получил название вестерн- блоттинг).
9.3. Строение генетических конструкций
Описанная технология рекомбинантных ДНК позволяет создавать генетические конструкции, которые содержат один или несколько так называемых целевых генов (генов, кодирующих определенный новый признак, который исследователь собирается придать организму), а также все необходимые регуляторные элементы, обеспечивающие активность (экспрессию) этих генов после их переноса в организмы. Кроме того, в целях упрощения отбора трансформированных клеток в генетические конструкции включают также последовательности, кодирующие селективные или репортерные гены с соответствующими регуляторными элементами. Рассмотрим эти составляющие генно-инженерных конструкций подробнее.
9.3.1. Целевые гены. Трансгены и цисгены. Смысловые и антисмысловые конструкции
Перечень официально разрешенных к хозяйственному использованию трансгенных сельскохозяйственных и декоративных культур с указанием привнесенных признаков и генов, которые их кодируют (по состоянию на август 2014 г.), представлен в табл. 9.2. Основные признаки: толерантность к гербицидам, устойчивость к насекомым-вредителям, устойчивость к вирусным болезням, удлинение сроков созревания и способность к длительному хранению, улучшенные качественные характеристики (состава жирных кислот растительного масла, крахмала, пониженное содержание никотина в листьях табака), улучшенные кормовые свойства, система получения гетерозисных гибридов на основе мужской стерильности/восстановления фертильности, устойчивость к засухе, пригодность для производства биотоплива. Отдельные признаки, например толерантность к гербицидам, можно конкретизировать в зависимости от гербицида: толерантность к глифосату, глюфозинату аммония, имидозолинону, сульфонилмочевине и т. д; устойчивость к насекомым: колорадскому жуку, повреждающему картофель, личинкам мотыльков (европейского точильщика кукурузы, хлопкового коробочного червя, розового коробочного червя хлопчатника и др.), корневым червецам на кукурузе и т. д. Имеется большое количество других ценных для селекции растений выделенных, клонированных и изученных генов, которые предполагается использовать для улучшения растений с помощью генно-инженерных технологий. Постоянно идет поиск новых генов, представляющих практический интерес, для чего активно используются эффективные методы геномики, протеомики, метаболомики, системной биологии и биоинформатики.
Как видно из табл. 9.2, основная масса генов кодирует названные признаки, бактериального или вирусного происхождения. Их называют трансгенами. К трансгенам также относят гены от неродственных по отношению к трансформируемому растений, с которыми практически невозможно получить половые или соматические гибриды, например, от арабидопсиса как источника гена толерантности к ALS-гербицидам для сои. Использование в генно-инженерных исследованиях чужеродных генов, которые ранее не применялись в селекции растений, явилось одной из причин возникновения сомнений относительно безопасности ГМО для здоровья человека и окружающей среды. В связи с этим предложено использовать в качестве целевых так называемые цис-гены: гены и их собственные регуляторные элементы от растений того же или близкородственного дикого вида, с которым возможна половая гибридизация (E. Jacobsen, H. Schouten, 2008).
Таблица 9.2. Перечень допущенных к использованию в хозяйственной деятельности трансгенных сортов сельскохозяйственных растений
(по CERA(2014). GM Crop Database (http:/cera-gmc.org))
Название культуры |
Количество трансгенных «событий» |
Фенотипический признак, встроенные гены |
Люцерна |
2 |
Толерантность к гербициду глифосату благодаря инсерции модифицированного гена epsps от Agrobacterium tumefaciensCP4 |
Рапс аргентинский |
3 |
Толерантность к фосфинотрициновым гербицидам (глюфозинату аммония) благодаря инсерции гена фосфинотрицин N-ацетил трансферазы от Streptomyces viridochromogenes |
Рапс аргентинский |
1 |
Толерантность к оксиниловым гербицидам, включая бромоксинил и иоксинил, благодаря инсерции гена нитрилазы от Klebsiella pneumoniae var. ozanae |
Рапс аргентинский |
2 |
Толерантность к гербициду глифосату благодаря инсерции модифицированного гена epsps от Agrobacterium tumefaciensCP4 и гена глифосат оксидазы от Ochrobactrum anthropi |
Рапс аргентинский |
1 |
Модифицированное содержание жирных кислот в семенах, особенно высокие уровни лаурата и образования миристиновой кислоты благодаря инсерции гена тиоэстеразы калифорнийского лаврового дерева |
Рапс аргентинский |
6 |
Система контроля опыления: мужская сте- рильность/восстановление фертильности благодаря инсерции гена barnase(стерильность) или гена barstar (восстановление фертильности) от Bacillus amyloliquefaciens; толерантность к фосфинотрициновым гербицидам (глюфозинату аммония) благодаря инсерции гена фосфинотрицин N-ацетил трансферазы от Streptomices hygroscopicus |
Гвоздика |
1 |
Увеличенный срок хранения благодаря сниженному накоплению этилена из-за введения усеченного гена аминоциклопропан циклаза синтазы гвоздики; толерантность к сульфонилмочевинным гербицидам (триа- сульфурону и метсульфурон-метилу) благодаря инсерции гена ацетолактат синтазы от линии табака Nicotiana tabacum, устойчивой к хлорсульфурону |
Гвоздика |
10 |
Модификация окраски цветка (фиолето- вый/розовато-лиловый) благодаря инсерции двух генов биосинтеза антоциана от Petunia hybrida; толерантность к сульфонилмочевинным гербицидам (триасульфурону и метсульфурон-метилу) благодаря инсерции гена ацетолактат синтазы от линии табака Nicotiana tabacum, устойчивой к хлорсульфурону |
Цикорий |
3 |
Система контроля опыления: мужская сте- рильность/восстановление фертильности благодаря инсерции гена barnase(стерильность) или гена barstar (восстановление фертильности) от Bacillus amyloliquefaciens; толерантность к фосфинотрициновым гербицидам (глюфозинату аммония) благодаря инсерции гена фосфинотрицин N-ацетил трансферазы от Streptomices hygroscopicus |
Хлопчатник |
1 |
Толерантность к оксиниловым гербицидам, включая бромоксинил и иоксинил, благодаря инсерции гена нитрилазы от Klebsiella pneumoniae |
Хлопчатник |
3 |
Толерантность к гербициду глифосату благодаря инсерции модифицированного гена epsps от Agrobacterium tumefaciensCP4 или мутантного гена epsps от кукурузы |
Хлопчатник |
1 |
Толерантность к фосфинотрициновым гербицидам (глюфозинату аммония) благодаря инсерции гена фосфинотрицин N-ацетил трансферазы от Streptomices hygroscopicus |
Хлопчатник |
6 |
Устойчивость к чешуекрылым насекомым (мотылькам) благодаря инсерции гена cry1Ac или cry1Ab от Bacillusthuringiensis sub- sp. kurstaki, или гена cry1F от Bacillus thuringiensis var. aizavai, или гена vip3A(a) от Bacillus thuringiensisАВ88 |
Хлопчатник |
1 |
Устойчивость к чешуекрылым насекомым (мотылькам) благодаря инсерции генов cry1Ac и cry2Ab от Bacillus thuringiensissub- sp. kurstaki |
Хлопчатник |
1 |
Устойчивость к чешуекрылым насекомым (мотылькам) благодаря инсерции гена cry1Ac от Bacillus thuringiensis subsp. kurstaki; толерантность к оксиниловым гербицидам, включая бромоксинил, благодаря инсерции гена нитрилазы от Klebsiella pneumoniae |
Хлопчатник |
1 |
Устойчивость к чешуекрылым насекомым (мотылькам) благодаря инсерции генов cry1Ac и cry2Ab от Bacillus thuringiensissubsp. kurstaki; толерантность к фосфинотрициновым гербицидам (глюфозинату аммония) благодаря инсерции гена фосфинотрицин N-ацетил трансферазы от Streptomices hygro- scopicus |
Хлопчатник |
7 |
Гибриды, полученные от скрещивания линий с трансгенами устойчивости к чешуекрылым насекомым (мотылькам) и линий с трансгенами толерантности к гербициду глифосату или гербициду глюфозинату аммония |
Полевица Agrostis stolonifera |
1 |
Толерантность к гербициду глифосату благодаря инсерции модифицированного гена epsps от Agrobacterium tumefaciensCP4 |
Лен |
1 |
Толерантность к сульфонилмочевинным гербицидам (триасульфурону и метсульфурон-метилу) благодаря инсерции гена ацетолактат синтазы от линии арабидопсиса Arabidopsis thaliana, устойчивой к хлорсульфурону |
Кукуруза |
3 |
Толерантность к гербициду глифосату благодаря инсерции модифицированного гена epsps кукурузы или гена глифосат оксидазы от Ochrobactrum anthropi |
Кукуруза |
3 |
Толерантность к фосфинотрициновым гербицидам (глюфозинату аммония) благодаря инсерции гена фосфинотрицин N-ацетил- трансферазы от Streptomices hygroscopicus или Streptomyces viridochromogenes |
Кукуруза |
1 |
Толерантность к гербициду глифосату благодаря инсерции модифицированного гена глифосатацетилтрансферазы от Bacillus licheniformis; и толерантность к сульфонил- мочевинным гербицидам благодаря инсерции модифицированного гена ацетолактат синтазы кукурузы |
Кукуруза |
3 |
Устойчивость к европейскому кукурузному точильщику (мотыльку Ostrinia nubilalis) благодаря инсерции гена cry 1Ab от Bacillus thuringiensis subsp. kurstaki |
Кукуруза |
2 |
Устойчивость к кукурузному корневому червю (чешуекрылые, виды Diabmtica sp.) благодаря инсерции модифицированного гена cry 3A от Bacillus thuringiensis subsp. tenebrionis или гена cry 3Bb1 от Bacillus thuringiensis subsp. kumamotoensis |
Кукуруза |
1 |
Устойчивость к комплексу насекомых-вре- дителей благодаря инсерции химерного гена cry1A.105 и гена cry 2Ab.2 |
Кукуруза |
3 |
Устойчивость к европейскому кукурузному точильщику (мотыльку Ostrinia nubilalis) благодаря инсерции гена cry lAb от Bacillus thuringiensis subsp. kurstaki; толерантность к гербициду глифосату благодаря инсерции модифицированного гена epsps от Agrobacterium tumefaciens CP4 |
Кукуруза |
5 |
Устойчивость к европейскому кукурузному точильщику (мотыльку Ostrinia nubilalis) благодаря инсерции генов cry lAb, или cry9C, или cry 1F, или vip 3A от Bacillus thuringiensis; толерантность к фосфинотрициновым гербицидам (глюфозинату аммония) благодаря инсерции гена фосфинотрицин N-ацетил- трансферазы от Streptomices hygroscopicus |
Кукуруза |
1 |
Устойчивость к кукурузному корневому червю благодаря инсерции генов cry 34Ab1 или cry 35Ab1 от Bacillus thuringiensisPS149B1; толерантность к фосфинотрициновым гербицидам (глюфозинату аммония) благодаря инсерции гена фосфинотрицин N-ацетил- трансферазы от Streptomyces viridochromogenes |
Кукуруза |
1 |
Устойчивость к кукурузному корневому червю благодаря инсерции гена cry 3Bbl от Bacillus thuringiensissubsp.kumamotoensis; толерантность к гербициду глифосату благодаря инсерции модифицированного гена epsps от Agrobacterium tumefaciens CP4 |
Кукуруза |
3 |
Мужская стерильность благодаря инсерции гена ДНК аденин метилазы от E. coli; толерантность к фосфинотрициновым гербицидам (глюфозинату аммония) благодаря инсерции гена фосфинотрицин N-ацетил- трансферазы от Streptomices hygroscopicus |
Кукуруза |
2 |
Мужская стерильность благодаря инсерции гена barnase от Bacillus amyloliquefaciens; толерантность к фосфинотрициновым гербицидам (глюфозинату аммония) благодаря инсерции гена фосфинотрицин N-ацетил- трансферазы от Streptomices hygroscopicus |
Кукуруза |
1 |
Повышенное содержание аминокислоты лизина в белке зерна благодаря инсерции гена дигидродипиколинат синтазы от Corynebacterium glutamaticum |
Кукуруза |
1 |
Кукуруза для производства спирта, содержащая в зерне фермент устойчивую к высоким температурам альфа-амилазу, благодаря инсерции гена amy от Thermococcales spp |
Кукуруза |
1 |
Повышенная устойчивость к засухе благодаря инсерции гена cspB протеина холодового шока от Bacillus subtilis |
Кукуруза |
1 |
Гибриды между линиями с трансгеном толерантности к гербициду глифосату и толерантности к гербициду глюфозинату аммония |
Кукуруза |
21 |
Гибриды между линиями с трансгенами устойчивости к насекомым и толерантности к гербицидам глифосату или глюфозинату аммония |
Кукуруза |
1 |
Гибрид между линией с трансгеном повышенного содержания лизина и линией с трансгеном устойчивости к насекомым |
Дыня |
2 |
Удлинение сроков созревания благодаря встраиванию гена фермента S-аденозилметионин гидролазы от бактериофага T3 E. coli, который приводит к деградации предшественника растительного гормона этилена |
Папайя |
2 |
Устойчивость к вирусу кольцевой пятнистости папайи (PRSV) благодаря инсерции гена белка оболочки (CP) PRSV |
Слива |
1 |
Устойчивость к покс-вирусу сливы (PPV) благодаря инсерции гена белка оболочки (CP) PPV |
Рапс польский (турнепс) |
1 |
Толерантность к гербициду глифосату благодаря инсерции модифицированного гена epsps от Agrobacterium tumefaciensCP4 |
Рапс польский (турнепс) |
1 |
Толерантность к фосфинотрициновым гербицидам (глюфозинату аммония) благодаря инсерции гена фосфинотрицин N-ацетил- трансферазы от Streptomyces viridochromogenes |
Картофель |
14 |
Устойчивость к колорадскому жуку (Leptinotarsa decemlineata) благодаря инсерции модифицированного гена cry 3A от Bacillus thuringiensis subsp. Tenebrionis |
Картофель |
3 |
Устойчивость к колорадскому жуку (Leptinotarsa decemlineata) благодаря инсерции модифицированного гена cry 3A от Bacillus thuringiensis subsp. Tenebrionis; устойчивость к (лютео)вирусу скручивания листьев картофеля, (PLRV) благодаря инсерции гена ре- пликазы PLRV |
Картофель |
3 |
Устойчивость к колорадскому жуку (Leptinotarsa decemlineata) благодаря инсерции модифицированного гена cry 3A от Bacillus thuringiensis subsp. Tenebrionis; устойчивость к Y вирусу картофеля (PVY) благодаря инсерции гена белка облочки (CP) PVY |
Картофель |
1 |
Измененный состав крахмала с увеличенным отношением ветвистой формы амилопектина к неветвистой амилозе благодаря инсерции гена картофеля gbss (связанной с гранулами синтетазы крахмала) в антисмысловой ориентации |
Рис |
3 |
Толерантность к фосфинотрициновым гербицидам (глюфозинату аммония) благодаря инсерции гена фосфинотрицин N-ацетил- трансферазы от Streptomices hygroscopicus |
Соя |
2 |
Толерантность к гербициду глифосату благодаря инсерции модифицированного гена epsps от Agrobacterium tumefaciensCP4 |
Соя |
7 |
Толерантность к фосфинотрициновым гербицидам (глюфозинату аммония) благодаря инсерции гена фосфинотрицин N-ацетил- трансферазы от Streptomices viridochromogenes или от Streptomices hygroscopicus |
Соя |
1 |
Толерантность к гербициду имидозолино- ну благодаря инсерции модифицированного гена csr1-2 от Arabidopsis thaliana |
Соя |
1 |
Толерантность к гербицидам глифосату и имдозолинону благодаря инсерции химерного гена глифосат N-ацетилтрансферазы от Bacillus licheniformis и модифицированного гена ацетолактат синтазы сои |
Соя |
4 |
Модификация содержания жирных кислот в семенах, особенно высокая экспрессия олеиновой кислоты благодаря инсерции дополнительной копии гена десатуразы сои |
Кабачки |
1 |
Устойчивость к вирусу 2 мозаики арбуза (WMV) и вирусу желтой мозаики цуккини (ZYMV) благодаря инсерции последовательностей ДНК протеинов оболочки (CP) каждого из этих вирусов |
Кабачки |
1 |
Устойчивость к вирусу мозаики огурцов (CMV), вирусу мозаики арбуза (WMV), вирусу желтой мозаики цуккини (ZYMV) благодаря инсерции последовательностей ДНК протеинов оболочки (CP) каждого из этих вирусов |
Сахарная свекла |
1 |
Толерантность к фосфинотрициновым гербицидам (глюфозинату аммония) благодаря инсерции гена фосфинотрицин N-ацетилрансферазы от Streptomices viridochromogenes |
Сахарная свекла |
2 |
Толерантность к гербициду глифосату благодаря инсерции модифицированного гена epsps от Agrobacterium tumefaciensCP4 |
Табак |
1 |
Толерантность к оксиниловым гербицидам, включая бромоксинил и иоксинил, благодаря инсерции гена нитрилазы от Klebsiella pneumoniae var. ozanae |
Табак |
1 |
Низкое содержание никотина благодаря инсерции дополнительной копии гена фосфорибозилтрансферазы табака в антисмысловой ориентации |
Томаты |
1 |
Удлинение сроков созревания благодаря инсерции гена фермента S-аденозилметионин гидролазы от бактериофага T3 E.coli; этот фермент деградирует предшественника растительного гормона этилена |
Томаты |
1 |
Удлинение сроков созревания благодаря инсерции гена деаминазы 1-амино- циклопропан-1-карбоновой кислоты от Pseudomonas chlororaphis; этот фермент деградирует предшественника растительного гормона этилена |
Томаты |
1 |
Удлинение сроков созревания благодаря инсерции дополнительной копии гена томатов синтазы 1-аминоциклопропан-1- карбоновой кислоты, что приводит к снижению аккумуляции растительного гормона этилена |
Томаты |
4 |
Удлиненный период хранения благодаря инсерции дополнительной копии гена полигалактуроназы томатов в смысловой или антисмысловой ориентации. Плоды дольше сохраняют упругость благодаря подавлению активности фермента полигалактуроназы, расщепляющего пектин |
Томаты |
1 |
Устойчивость к чешуекрылым насекомым (мотылькам) благодаря инсерции гена cry1Ac от Bacillus thuringiensis subsp. kurstaki |
Пшеница |
1 |
Толерантность к гербициду глифосату благодаря инсерции модифицированного гена epsps от Agrobacterium tumefaciensCP4 |
В целях изменения активности отдельных генов растений, связанных с контролем качественных характеристик или сроков созревания плодов, применяют перенос генетических конструкций, содержащих кодирующие последовательности самих этих генов (т. е. генов того же вида растения). Вставка дополнительной копии гена во многих случаях приводит к его замолканию (явление посттранскрипционного замолкания гена - англ. posttranscriptional gene silencing) или существенному снижению активности. Дополнительные копии генов вводят в смысловой или антисмысловой ориентации (перевернутой концом по отношению к промотору, в результате чего образуется мРНК, комплементарная нормальной мРНК гена). В основе механизма замолкания гена образование двунитчатых мРНК, которые расщепляются в клетке на мелкие фрагменты (англ. small interfering RNAs - si-RNA). Последние, соединяясь со специфическими протеинами, образуют так называемый РНК- индуцированный комплекс замолкания (англ. RNA-induced silencing complex), который способен вызывать деструкцию гомологичных мРНК в ядре клетки (H. Yan и др., 2006).
Использование для целей генетической модификации исключительно собственных генов растений лежит в основе интрагенного подхода при получении ГМО (C. Rommens и др., 2004). Применяя различные тканеспецифические промоторы (см. 9.3.4), можно добиться ослабления или усиления экспрессии отдельных генов и благодаря этому улучшить качественные характеристики существующих сортов.
9.3.2. Селективные и репортерные гены. Получение трансгенных растений без селективных генов
Поскольку процесс трансформации затрагивает лишь небольшое количество компетентных клеток, то для их отбора используют селективные маркерные гены, которые, как правило, присутствуют в переносимых в растительные клетки генетических конструкциях наряду с генами целевых признаков. При высеве подвергнутых трансформации клеток на питательную среду, содержащую селективный агент, способностью делиться будут обладать только те клетки, в геном которых произошла вставка рекомбинантной ДНК. В генетической инженерии растений чаще всего в качестве маркерных используют гены устойчивости к антибиотикам и гены устойчивости к гербицидам. Если целью генетической модификации является получение гербицидоустойчивых форм, то сам целевой ген может выступать в качестве селективного гена.
Из селективных генов устойчивости к антибиотикам наиболее широкое применение нашел ген nptII (синонимы neo, aphII) из транспозона Tn5 E. coli, кодирующий фермент неомицинфосфотрансферазу II. Этот фермент катализирует присоединение к аминогликозидным антибиотикам канамицину, неомицину, гентамицину остатка фосфорной кислоты, в результате чего происходит их дезактивация. Популярность этого гена обусловлена тем, что он очень эффективен, пригоден для широкого круга видов растений и хорошо изучен. Его экспрессия в растении не влияет на экспрессию растительных генов. Многочисленные исследования, а также уже достаточно длительная история его использования показали, что продукт этого гена является безопасным для здоровья человека и состояния окружающей среды. Он присутствует в генетических конструкциях ряда трансгенных растений, официально допущенных к использованию в качестве продуктов питания и кормов. Кроме гена nptII в генно-инженерных исследованиях фигурируют также гены устойчивости к таким антибиотикам, как гигромицин В, стрептомицин, хлорамфеникол и др.
Среди генов устойчивости к гербицидам чаще всего используют ген фосфинотрицин N-ацетилтрансферазы (PAT) - фермента, дезактивирующего фосфи- нотрицин (глюфозинат аммония), который является активным компонентом широко применяемого во всем мире гербицида фирмы «Bayer». Дезактивация гербицида происходит в результате его ацетил-CoA ацетилирования с помощью РАТ. На практике нашли применение два гена, кодирующих этот фермент: ген bar устойчивости к биалофосу (биалофос содержит фосфинотрицин и два остатка L-аланина) от Streptomyces hygroscopicus и ген pat от Streptomyces viridochromogenes. Эти гены особенно эффективны применительно к злаковым растениям, поскольку канамицин для однодольных недостаточно токсичен.
Хотя, как показывает практика и результаты специальных исследований, наличие в трансгенных конструкциях селективных генов устойчивости к антибиотикам и гербицидам не является опасным для здоровья человека и окружающей среды, но, учитывая озабоченность, а часто и неприятие общественностью этого факта, ученые прилагают усилия по разработке альтернативных селективных систем. В частности, предложено использовать нетоксичные источники углерода, которые не могут быть утилизированы растительными клетками, если они не имеют фермента, способного их катаболизировать. Примером таких селективных агентов являются манноза, D-ксилоза, 2-деоксиглюкоза. Имеется ряд бактериальных генов, кодирующих ферменты, необходимые для включения этих сахаров в гликолиз. Это, в частности, ген man фосфоманноза изомеразы от E. coli, ген xylA ксилоза изомеразы от Streptomyces rubiginosus или Thermoanaerobacterium thermosulfurogenes. Селективные системы на основе нетоксичных сахаров показали высокую эффективность применительно к широкому кругу видов растений. Их действие более мягкое по сравнению с антибиотиками или гербицидами, что обеспечивает более высокую частоту трансформации.
Еще один, сравнительно новый тип селективной системы основан на использовании генов, обеспечивающих ростовые преимущества или повышенную способность к морфогенезу трансформированных клеток по сравнению с нетрансформированными. В частности, для этих целей нашел применение ген ipt изопентил трансферазы из Ti-плазмиды A. tumefaciens,который кодирует биосинтез предшественника цитокинина изопентинил аденозин- 5'-монофосфата. Экспрессия этого гена в трансформированных клетках повышает частоту стеблевой регенерации. При этом в связи с избыточным биосинтезом цитокининов возможны проблемы с укоренением растений- регенерантов. Для их решения предложено использовать индуцибельный промотор, позволяющий сократить время экспрессии гена ipt.
Имеется специфическая группа маркерных генов, которая широко используется в генетической инженерии растений. Речь идет о так называемых репортерных генах, которые способны менять внешний вид трансформированных клеток, благодаря чему их можно визуально отличить от нетрансформированных. Репортерные гены помещают в генетических конструкциях вслед за промотором впереди последовательности, кодирующей целевой ген. Благодаря этому по степени экспрессии репортерного гена (ее можно оценить количественно) можно судить и об экспрессии целевого гена. К репортерным генам и их продуктам предъявляют следующие требования: они не должны иметь фоновой активности (т. е. практически отсутствовать у интактных растений); не должны оказывать значительного влияния на метаболизм клетки хозяина; должны оставаться активными при значительных достройках белка; быть небольшого размера; иметь простые, чувствительные методы количественного и качественного определения белка. Репортерные гены применяют для изучения регуляторных элементов: промоторов, энхансеров, сайлэн- серов, терминальных последовательностей. В частности, при исследовании промоторов можно определить области, связанные с тканеспецифической экспрессией генов, или выделить у них светочувствительные участки и т. д.
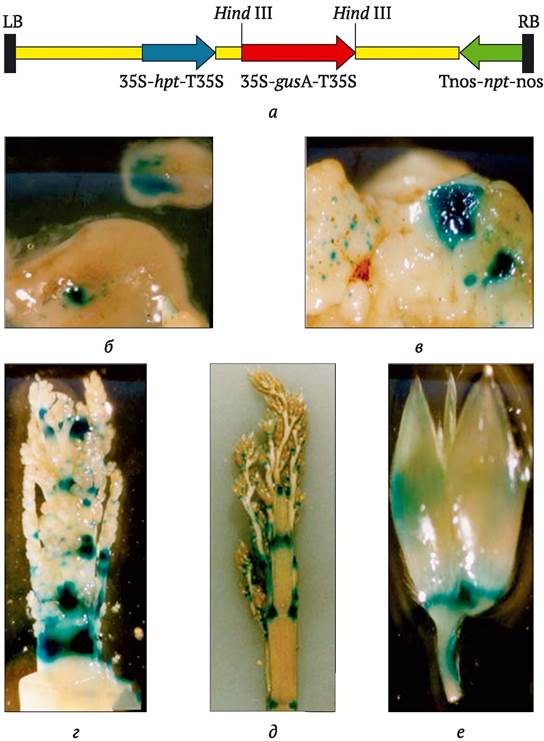
Чаще всего в качестве репортерного гена используют ген gus (или uidA) из E. coli, кодирующий образование фермента ß-глюкоронидазы (рис. 9.8). Экспрессию этого гена оценивают с помощью спектрометрических или гистохимических методов. В присутствии субстрата 4-метилумбеллиферилглюкоро- нида (MUG) появляется синяя окраска, интенсивность которой коррелирует со степенью экспрессии гена gus. Для выявления трансформированных клеток гистохимическими методами (они также приобретают синюю окраску) используют 5-бромо-4-хлоро-3-индолил глюкуронид (X-gluc). У интактных (нетрансформированных) растений продукт этого гена практически не выявляется. Фермент стабилен в растительных клетках и может накапливаться в значительных количествах без какой-либо токсичности. В отсутствие субстрата фермента трансформированное растение не отличается от интактного. Сам анализ простой и высокочувствительный. Среди его недостатков - необходимость разрушения растительных тканей трансформантов для проведения исследований. Поэтому в тех случаях, когда необходимо проследить динамику экспрессии трансгена в онтогенезе растения, рекомендуют использовать такие гены, как ген люциферазы (luc) из светлячков Photinus pyralis или ген gfp зеленого флюоресцентного протеина (GFP - green fluorescent protein) из медузы Aequorea victoria.
Рис. 9.8. Пример использования репортерной системы GUS при трансформации сорго: а - схема бинарного вектора pTOK233, включающего, помимо репортерного гена β-глюкоронидазы (gusA), также два селективных гена устойчивости к антибиотикам гигромицину hpt и канамицину npt; б-е - экспрессия встроенного гена gusA: б - в клетках незрелого эмбриоида сорго; в - в клетках каллюсной ткани, формирующейся из незрелого эмбриоида; 2 и д - в развивающихся соцветиях сорго на разных стадиях зрелости; е - в цветке сорго на стадии созревания пыльцы (наибольшая экспрессионная активность гена наблюдается в тканях с активным ростом клеток) (по C. Carvalho и др., 2004)
Фермент люцифераза катализирует АТФ-зависимое оксидативное декарбоксилирование субстрата люциферина. Экспрессия гена люциферазы придает трансформированным клеткам способность к биолюминесценции. Экспрессия зеленого флюоресцентного протеина (GFP) не связана с каким- либо субстратом: флюоресценцию трансформированных клеток наблюдают в ультрафиолетовом свете, ее можно оценить количественно. GFP нетоксичен, устойчив к действию протеиназ и повышенной температуры, оказывает минимальное влияние на подвижность и локализацию гибридного белка (GFP, соединенного с целевым протеином под одним промотором).
GFP впервые был выделен и изучен О. Shimomura c сотрудниками (1962). Однако применение GFP в молекулярной биологии началось лишь в 1990-е гг. после публикации работ D. Prasher по клонированию и секвенированию кодирующей последовательности этого гена. Позднее с помощью мутагенеза были значительно улучшены спектральные характеристики GFP и его физико-химические свойства (чувствительность к изменению pH, фото- и термостабильность). В частности, благодаря точковой мутации S65T (R. Heim и др., 1995), была усилена флюоресценция GFP, его фотостабильность, произошел сдвиг основного пика активации к 488 нм с пиком эмиссии свечения при 509 н. м., что хорошо подходило к спектральным характеристикам обычно используемых флюоресцентных фильтров и, как следствие, обеспечило возможность использования GFP практически в любой лаборатории. Были получены мутантные формы GFP, а также выделены другие флюоресцентные белки с синим, голубым, красным, оранжевым и желтым свечением. Их использование позволяет наблюдать одновременно экспрессию нескольких генов. Репортерная система на основе GFP нашла широкое применение не только в генно-инженерных исследованиях, но и других областях знаний. Первооткрыватель GFP О. Shimomura совместно с M. Chalfie и R. Tsien были удостоены Нобелевской премии по химии за 2008 г.
С практической точки зрения маркерные гены в трансгенных растениях не выполняют какой-либо роли. Они необходимы лишь на стадии получения ГМО для отбора трансформированных клеток. Помимо проблем, связанных с негативным отношением общественности к присутствию маркерных генов у ГМО, они могут создавать определенные неудобства в работах по совершенствованию трансгенных сортов, так как при каждом последующем цикле модификации необходимо использовать новые селективные системы, тем самым расширяя круг «ненужных» растению генов. Следует также иметь в виду, что применение селективных систем, основанных на толерантности к нетоксичным сахарам, повышенной регенерационной способности трансформированных клеток, может существенно затрагивать метаболизм ГМО, что нежелательно с точки зрения биобезопасности. В связи с этим были разработаны методы получения трансгенных растений без таких генов, а также методы удаления селективных генов у трансформантов после проведения селективной процедуры.
В тех случаях, когда эффективность трансформации достаточно высока, можно использовать трансгенные конструкции, не содержащие маркерные гены. Отобрать трансгенные генотипы (фенотипы) несложно с помощью детекции целевого гена у всех полученных растений-регенерантов или их исследования на наличие привнесенного признака. Целевой ген толерантности к гербицидам может быть использован и в качестве селективного для отбора трансформированных клеток.
Эффективным методом получения безмаркерных ГМО является котрансформация с последующим негативным отбором по селективным генам в расщепляющихся поколениях полученных трансформантов. Предложено несколько способов котрансформации. Например, целевой и селективный гены могут располагаться на разных плазмидах одного штамма агробактерии или на плазмидах разных штаммов, которыми одновременно инфицируют растительные ткани (процесс агробактериальной трансформации описан в разделе 9.5). Эти гены могут быть на одной плазмиде, но на разделенных пространственно участках ее ДНК, чтобы в половых поколениях ГМО эти гены сегрегировали независимо. При высокой частоте трансформации имеется возможность отобрать трансгенные линии, несущие одновременно как селективный, так и целевой ген. Анализируя самоопыленное потомство таких ГМО или беккроссы на исходный сорт, проводят отбор генотипов, имеющих только целевой ген. Метод котрансформации имеет ряд ограничений. В частности, он может быть использован в основном к размножаемым семенами самоопылителям, представляющим собой относительно гомозиготные линии, и не пригоден для вегетативно размножаемых культур, которые по большей части гетерозиготы. Также необходимо наличие протокола трансформации культуры, обеспечивающего высокий выход трансгенных генотипов.
Для удаления селективных генов из генома ГМО используют генетические конструкции, содержащие последовательности транспозонов. В частности, описана система с селективным маркерным геном ipt изопентилтрансферазы, соединенным с транспозонным элементом Ас. Первоначально ген ipt выступает в качестве позитивного селективного агента, вызывая образование многочисленных стеблевых отростков, из которых не могут быть регенерированы нормальные трансгенные растения из-за избытка цитокининов. Однако через несколько недель культивирования с небольшой частотой появляются стебли с нормальным фенотипом. Это происходит благодаря транспозиции гена ipt и его сегрегации в соматических клетках (H. Ebinuma и др., 2001).
Второй подход по удалению селективных генов у ГМО основан на использовании сайт-специфических рекомбиназ. Известны рекомбиназы и последовательности ДНК, которые могут быть мишенями этих ферментов. Это, например, Cre/lox из бактериофага Р1, FLP/FRT из дрожжей Saccharomyces cerevisiae, R/RS из Zygosaccharomyces rouxii. Эти последовательности размещают в генетической конструкции по краям гена, который планируется позднее удалить. Затем во втором туре трансформации вводят ген рекомбиназы. Последний ген, после того как он выполнил свою функцию, удаляют путем негативного отбора в беккроссном поколении ГМО (расщепление по этому гену и целевому гену происходит независимо). Возможен также вариант транзиентной экспрессии гена рекомбиназы. Удаление селективного гена происходит с более низкой частотой, однако при этом ген рекомбиназы не встраивается в растительный геном и нет необходимости его затем удалять. Еще один перспективный подход - использование генетических конструкций с целевым геном, селективным геном и геном рекомбиназы на одном векторе. Ген рекомбиназы помещают под индуцибельный промотор, благодаря чему в нужный момент можно запустить процесс одновременного самоудаления гена рекомбиназы и селективного гена (Н. Еbinuma и др., 2001).
9.3.3. Регуляторные элементы
Каждый ген имеет сложную систему регуляции своей активности, в отсутствие которой он не может функционировать. В частности, для транскрипции гена обязательно наличие, помимо кодирующей области, также промотора и терминальной последовательности (терминатора). Промоторы содержат специфические последовательности ДНК, включающие СААТ- и ТАТА-боксы, к которым присоединяется РНК-полимераза II, после чего начинается процесс транскрипции. Терминаторы содержат последовательности сигнала терминации транскрипции (у эукариот это обычно ГЦ-богатые последовательности), а также расположенной приблизительно на 30 п. н. выше последовательности сигнала полиаденилирования ААТАА, которая обеспечивает присоединение к мРНК поли-А-хвоста. Считается, что присоединение к 3'-концу мРНК по окончании транскрипции многочисленных остатков аденина (поли-А-хвоста) способствуют стабильности мРНК. В придачу к этим обязательным элементам генетические конструкции могут содержать также другие регуляторные элементы, определяющие, например, место и время активности переносимого гена.
Выбор промотора при создании трансгенных конструкций имеет особое значение. Существует общая закономерность: прокариотические промоторы могут обеспечить активность любого гена, в том числе и эукариотического, только в прокариотическом организме (у бактерий, синезеленых водорослей). В эукариотическом организме может функционировать только ген, имеющий эукариотический промотор. Поэтому при переносе генов от одного вида растений к другому можно использовать гены с их собственными промоторами (что характерно для цис-генеза или интрагенеза). Но если в растение переносится бактериальный ген, то промотор у него должен быть заменен на растительный. На практике и для растительных генов используют промоторы других генов, чтобы повысить их экспрессию или обеспечить их экспрессию в определенных органах растения или в определенный период онтогенеза.
В генетической инженерии растений широкое применение нашли промоторы от растительных вирусов, в частности, промотор CaMV35S вируса мозаики цветной капусты, промотор FVМ35S вируса мозаики норичника (Scrofularia ssp.). В процессе эволюции вирусы получили очень сильные промоторы, способные функционировать в любом генетическом окружении. Это свойство вирусных промоторов очень ценно, поскольку обеспечивает активную экспрессию привнесенного трансгена во всех клетках растения на протяжении всего онтогенеза. Такие промоторы называют конститутивными. Наличие сильного конститутивного промотора особенно важно для селективных маркерных генов, поскольку селективная процедура применяется как на клеточном уровне после проведения трансформации, так и на этапе регенерации и укоренения растений-регенерантов. Кроме того, ПЦР-детекция маркерного гена может быть эффективно использована для подтверждения инсерции трансгенов на организменном уровне, для анализа характера расщепления по привнесенным генам в половых поколениях трансформантов (см. ниже).
Безусловным лидером по частоте использования в генно-инженерных исследованиях является промотор CaMV35S малой субъединицы вируса мозаики цветной капусты (общепринятая аббревиатура 35 S-промотор). Среди часто используемых в генетической инженерии конститутивных промоторов также промотор гена nos нопалинсинтазы Agrobacterium tumefaciens (он немного слабее 35S-промотора). Эти промоторы пригодны для широкого круга видов двудольных растений. Для злаковых культур более эффективными конститутивными промоторами являются промоторы генов ractl актина риса и ZmUbi убиквитина кукурузы.
Если исследователь ставит более сложные задачи в плане регулирования активности трансгенов, то в его распоряжении имеется в настоящее время достаточно широкий выбор промоторов и других регуляторных элементов, комбинирование которых позволяет достичь тонкой регуляции активности трансгенов в пространстве (в определенных тканях растения) и времени (в определенный период развития). В качестве примеров тканеспецифических промоторов можно назвать: промотор PSsuAra из Arabidopsis thaliana, который обеспечивает экспрессию находящихся под ним трансгенов исключительно в зеленых тканях (листьях, стебле); промотор PTA29 из табака Nicotiana tabacum - в пыльниках, промотор гена gbss картофеля - в клубнях, промотор гена напина турнепса или промотор Kti3 гена ингибитора трипсина Куница сои - в семенах. Промотор гена rbcS малой субъединицы рибулезобифосфат карбоксилазы-оксигеназы Arabidopsis thaliana - светочувствительный: экспрессия расположенных под ним трансгенов на свету (например, в листьях) в сто и более раз выше, чем в темноте (например, в корнях, клубнях).
В качестве терминаторов в трансгенных конструкциях часто используют терминальные последовательности генов nos Agrobacterium tumefaciens и CaM- V35S малой субъединицы вируса мозаики цветной капусты, различных растительных генов (pinII ингибитора протеиназы картофеля, tahsp 17 белка теплового шока пшеницы, rbcS малой субъединицы фермента рибулезобифосфат карбоксилазы гороха, sphasl запасного белка сои конглицина, phas запасного белка фасоли фазеолина и др.), а также нативные терминальные последовательности переносимых растительных генов.
Кроме названных регуляторных элементов в генетических конструкциях могут присутствовать и другие специальные последовательности. Так, для того чтобы обеспечить эффективное включение протеинов-продуктов трансгенов в метаболизм клетки, трансгенные конструкции могут содержать, например, последовательности, кодирующие образование транзитного пептида, обеспечивающего доставку трансгенного фермента EPSPS к хлоропластам - месту синтеза ароматических аминокислот (генно-инженерная соя, устойчивая к гербициду глифосату).
Таким образом, для успешного переноса и стабильной, адресной экспрессии в организме-хозяине трансгенные конструкции должны иметь определенный набор кодирующих повторностей и соответствующих регуляторных генетических элементов. В качестве примера типичной трансгенной конструкции можно привести ту, что была использована при получении сортов рапса с системой мужской стерильности/восстановления фертильности, позволяющей получать гетерозисные гибридные семена (табл. 9.3). Между левым и правым краем от A. tumefaciens (которые, как известно, играют ключевую роль в переносе Т-области из агробактерии в растительную клетку, см. раздел 9.5) в этой конструкции расположены два гена со своими регуляторными элементами: ген целевого признака - мужской стерильности barnase и селективный маркерный ген bar толерантности к гербициду глюфозинату аммония (фосфинотрицину). Отдельные фрагменты ДНК соединены между собой с помощью синтетических полилинкеров.
Таблица 9.3. Состав трансгенной конструкции при создании ГМ-рапса с системой получения гибридных семян на основе мужской стерильности/восстановления фертильности и толерантности к гербициду глюфозинату аммония
Аббревиатура |
Генетический элемент |
Происхождение |
Размер п. н. |
Функция |
RB |
Правый край |
Agrobacterium riimefaciens |
25 |
Встраивание ДНК |
PLS |
Полилинкер |
Синтетический |
72 |
Соединение фрагментов ДНК |
3'g7 |
Сигнал терминации транскрипции от гена 7 TL-ДНК |
Agrobacterium tumefaciens |
212 |
Терминация транскрипции, присоединение поли-А |
PLS |
Полилинкер |
Синтетический |
21 |
Соединение фрагментов ДНК |
bar |
Ген толерантности к глюфозинату |
Streptomices hygroscopicus |
552 |
Селективный маркер |
PSsuAra |
Промотор |
Arabidopsis thaliana |
1726 |
Конститутивный промотор, экспрессия гена в зеленых тканях |
PLS |
Полилинкер |
Синтетический |
50 |
Соединение фрагментов ДНК |
3'nos |
Последовательность поли- аденилирования от гена nos |
Agrobacterium tumefaciens |
261 |
Присоединение поли-А |
3'UTR |
Сигнал терминации транскрипции от гена barnase |
Bacillus amyloliquefaciens |
112 |
Терминация транскрипции |
barnase* |
Ген РНКазы |
Bacillus amyloliquefaciens |
336 |
Мужская стерильность |
PTA29 |
Промотор |
Nicotiana tabacum |
1510 |
Конститутивный промотор, экспрессия гена в пыльниках |
PLS |
Полилинкер |
Синтетический |
44 |
Соединение фрагментов ДНК |
LB |
Левый край |
Agrobacterium riimefaciens |
25 |
Встраивание ДНК |
* У восстановителя фертильности на этом месте ген barstar (273 п. н.) - ингибитор РНКаз со своей собственной терминальной последовательностью (40 п. н.).
Интересно, что ген bar толерантности к гербициду глюфозинату аммония играет в этой конструкции тройную роль. Во-первых, он обеспечивает эффективную селекцию трансформированных клеток. Во-вторых, толерантность к гербициду - сам по себе важный агрономический признак. В-третьих, этот ген необходим в системе производства гибридных семян для размножения родительских линий.
9.4. Методы переноса генов в живые клетки
Выше в общих чертах была описана методика введения рекомбинантных плазмид в клетки бактерий Е. coli для клонирования генов. Однако клетки растений существенно отличаются от бактериальных по своей способности поглощать чужеродную ДНК. Прежде всего растительные клетки имеют жесткие целлюлозные оболочки, преодолеть которые весьма непросто. Поэтому для их трансформации используют специфические методы. В целях же обеспечения возможности применения обычных для микроорганизмов и животных клеток способов разработаны эффективные методы трансформации протопластов растительных клеток.
Для трансформации растительных протопластов применяют хорошо известные для бактерий методы стимуляции этого процесса с помощью CaCl2, а также полиэтиленгликоля. Очень эффективным методом оказалась и электропорация. Весьма перспективной рассматривается методика введения чужеродной ДНК в протопласты с помощью микроинъекций (рис. 9.9). Созданы компьютерные системы на основе соединенных с инвертированным микроскопом микроманипуляторов, обеспечивающие обнаружение, прикрепление и перемещение отдельных протопластов и микрокапилляров, через которые вводят ДНК в клетку. Однако главная проблема в трансформации протопластов растений - возможность использовать эту технологию лишь для ограниченного числа видов, для которых разработаны эффективные методы выделения, культивирования протопластов и получения из них растений-регенерантов.
Рис. 9.9. Микроинъекция ДНК в протопласты растений: а - автоматизированная универсальная система SMSR для микроинъекций в клетки (на схеме представлены основные блоки: микроскоп, снабженный двумя манипуляторами, движущимися в системе координат X, Y, Z, предметным рабочим столиком с нижней подсветкой, движущимся в системе координат X и Y и автоматизированной компьютерной системой управления); б - процесс осуществления микроинъекции ДНК (протопласт закреплен на кончике капилляра (справа), микроинъекция ДНК выполняется с помощью ультратонкого микрокапилляра (слева)); в - оператор в процессе работы (по H. Matsuoka и др. (http://www.intechopen.com/books/))
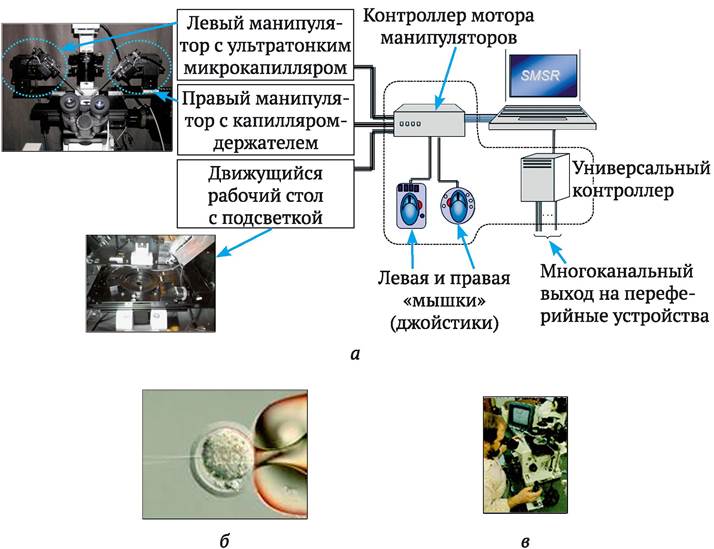
Среди методов прямого переноса генов в растительные клетки наибольшее распространение получил метод биологической баллистики (биолистики), или, как его еще называют, бомбардировки частицами (англ.: particle bombardment), генного дробовика (англ.: gene gun technique) (J. Sanford и др., 1987; T. Klein и др., 1987). Суть метода заключается в том, что на частицы диаметром 0,4-1,7 мкм из вольфрама или золота напыляют рекомбинантные ДНК, содержащие предназначенные для переноса гены с необходимыми регуляторными элементами. Эти частицы разгоняют до скорости 300-600 м/сек в специальном пневматическом аппарате - генной пушке (рис. 9.10) и помещают на их пути растительные ткани (меристемы, незрелые зародыши, колеоптили, протокормы, пыльцу) или культуры клеток (каллюс или суспензию клеток). В результате частицы проникают в клетки, а чужеродная ДНК встраивается в ДНК хромосом или органелл. Несмотря на ряд недостатков (нежелательная для трансформации растений многокопийность вставок, разрушение части вносимых генных конструкций и др.), метод биолистики - основной для получения трансгенных однодольных растений, среди которых все злаковые культуры, а также для многих двудольных растений, которые являются некомпетентными или слабо компетентными для основного метода введения генов в растительные клетки - агробактериальной трансформации.
Рис. 9.10. Оборудование, используемое для биобаллистической трансформации растений: а - переносная портативная генная пушка, используемая для трансформации in plantaпосредством обработки листьев; б - работа на стационарной генной пушке; в - схема строения генной пушки (http://www.ptf.okstate.edu/chambergun.html)
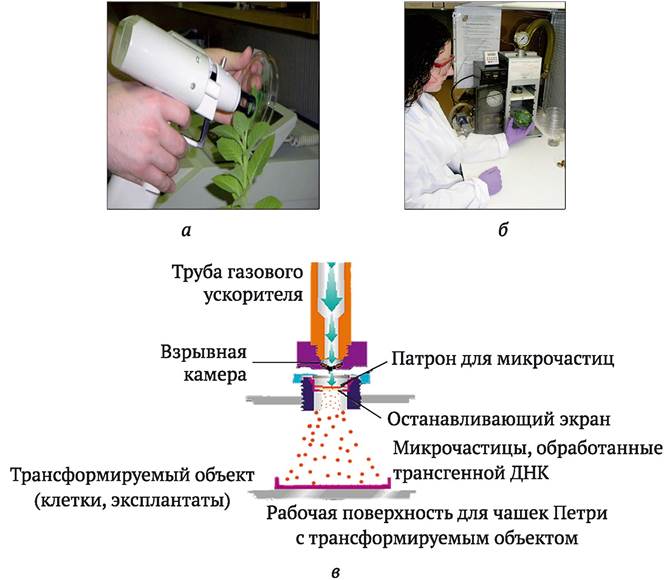
9.4.1. Агробактериальная трансформация
Раскрытие природы агробактериальной трансформации по праву считается одним из величайших научных достижений XХ в., поскольку дало возможность использовать в генетической инженерии уникальный природный механизм горизонтального переноса генов (т. е. переноса генов между отдаленными в систематическом отношении видами организмов).
Онкологическое заболевание растений - корончатый галл - издревле известно людям (его упоминание имеется еще в трудах Аристотеля). Этой болезни, имеющей серьезные агрономические последствия из-за нарушения нормального роста растений, подвержены многие двудольные растения: виноград, косточковые фруктовые деревья, розы и др. В начале прошлого века был установлен возбудитель заболевания - почвенные бактерии из рода Agrobacterium: A. tumefaciens (вызывает у растений образование опухоли - корончатого галла), A. rhizogenes (вызывает образование на стебле растений многочисленных корней (англ. hairy root), A. rubi (возбудитель галлов у тростника). Однако только в 1970-х гг. удалось раскрыть механизм патогенеза. N. van Larebeke с сотрудниками (1974), I. Zaenen c сотрудниками (1974) установили, что у вирулентных штаммов A. tumefaciens в отличие от невирулентных имеется большая плазмида, получившая название Ti (англ. tumor inducing - индуцирующая опухолеобразование) у A. tumefaciens. Подобная плазмида была выявлена и у вирулентных штаммов A. rhizogenes и получила название Ri (англ. root inducing - индуцирующая образование корней).
В ходе изучения этих плазмид выяснилось, что опухоль возникает в результате необратимого переноса и включения определенного фрагмента (Т-ДНК, англ. transferred DNA - переносимой ДНК) плазмиды в хромосомную ДНК растительной клетки (M.-D. Chilton и др., 1977). При этом гены, расположенные на Т-фрагменте ДНК бактериальной плазмиды, активно экспрессируются в растительной клетке, что выражается в синтезе большого количества специфических соединений опинов - октопинов и нопалинов при заражении A. tumefaciens или агропинов при заражении A. rhizogenes. Помимо этих соединений в клетках образуются в повышенных концентрациях различные фитогормоны, резко стимулирующие ростовые процессы, что приводит к образованию опухоли. Опины, экскретируемые инфицированными клетками, служат источником азота, углерода и энергии для возбудителей заболевания - агробактерий, поскольку у них на Ti-плазмиде имеются гены, кодирующие ферменты, необходимые для катаболизма этих соединений. Как видим, агробактерии создали для себя весьма уютную экологическую нишу, в которой у них практически нет конкурентов.
Для инициации процесса агробактериальной трансформации необходимо прежде всего, чтобы растение было повреждено, например, механическим путем при вспашке, прополке и т. п. Пораненные клетки выделяют в окружающую среду различные химические соединения, которые привлекают агробактерии (положительный хемотаксис). Далее бактерии прикрепляются к растениям в месте поранения с помощью целлюлозных фибрилл. Плотный контакт между бактериальной и растительной клетками является одним из важных факторов успешной трансформации. В генетическом контроле описанных начальных этапов агробактериальной трансформации в основном задействованы гены, расположенные на хромосоме бактерии. Их активность определяет все многообразие взаимодействий между бактерией и растением, включая распознавание растения-хозяина (определенные штаммы агробактерий могут трансформировать определенные виды растений) (рис. 9.11).
Рис. 9.11. Агробактериальная трансформация: а - внешний вид корончатого галла на стволе дерева; б - основные этапы агробактериальной трансформации (по B. Meyers и др., 2010)
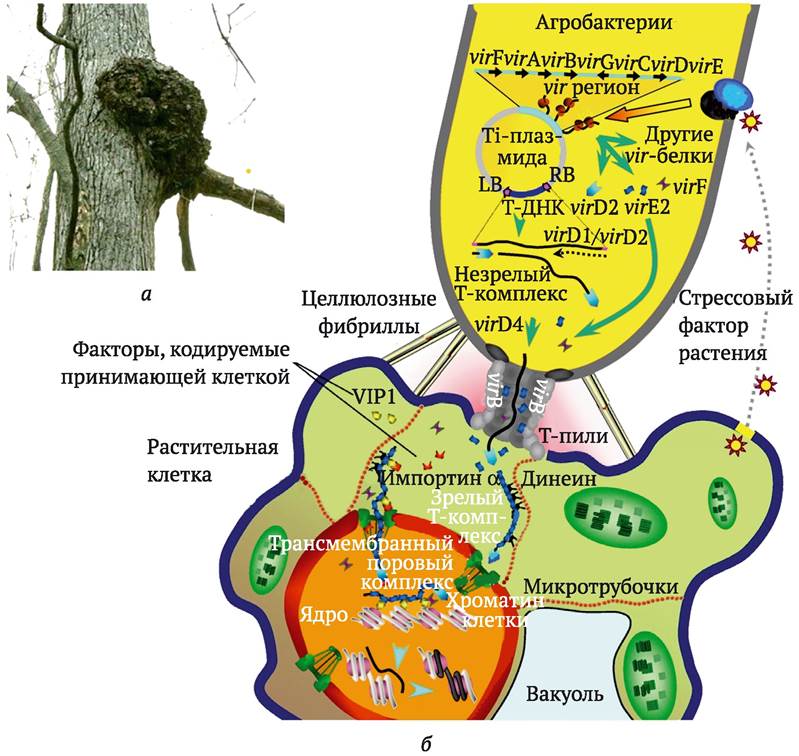
Однако в генетическом контроле собственно переноса Т-ДНК главными являются гены, расположенные в vir-области Ti-плазмиды (vir-гены плазмиды). Некоторые хромосомные гены могут влиять на активность плазмидных vir-генов. Плазмидные vir-гены расположены на одном регулоне размером около 40 т. п. н., который расположен отдельно от Т-области. У октопиновых штаммов агробактерий vz'r-регулон содержит не менее 10 идентифицированных vir-локусов, состоящих из нескольких генов, которые обозначают virA, virB и т. д. до virJ. У нопалиновых штаммов агробактерий в аналогичном регулоне обнаружено семь локусов.
Активность vir-генов индуцируется сигнальными молекулами химических соединений, выделяемых растениями при поранении: фенольными соединениями типа ацетосирингона и альфа-гидрооксиацетосирингона, предшественниками лигнина, синапиновой кислотой, кониферил алкоголем и др. или некоторыми флавоноидами. Для индукции vir-генов существенным является также наличие кислой среды, легко катаболизируемых источников углерода, например, сахарозы или моносахаридов, температуры ниже +30 °С.
Индуцирующий сигнал соединений, выделяемых растениями при поранении, воспринимается протеинами - продуктами генов virA и virG (англ.: signal-transduction complex). Протеин VirA является трансмембранным протеином, который имеет короткий периплазматический связывающий домен (место прикрепления определенных молекул) - N-терминал, воспринимающий сигналы от растительных фенольных соединений, и длинный, цитоплазматический район так называемый С-терминал. С-терминал обладает автофосфорилирующей активностью и способен активировать путем фосфорилирования протеин VirG. Активированный VirG - протеин, способный связываться с ДНК. Он распознает соответствующие регуляторные последовательности других плазмидных vir-генов и способен активировать их транскрипцию.
Активация экспрессии vir-генов под действием VirG приводит к запуску ряда последовательных взаимосвязанных событий, которые обеспечивают подготовку Т-ДНК к переносу в растительную клетку. Первый этап включает сайт-специфическое вырезание кодирующей нити Т-ДНК протеинами-продуктами генов virD (англ. helicase/endonuclease protein complex). Полипептид VirD2 в присутствии протеина VirDl распознает специфические последовательности Т-ДНК, состоящие из 25 п. н., расположенные в начале и конце Т-области, и производит вырезание кодирующей нити ДНК. Эти последовательности получили название левый (LB - left border) и правый (RB - right border) край Т-области соответственно. Они играют исключительно важную роль в процессе переноса Т-ДНК из бактериальной клетки в растительную.
Вырезанная кодирующая нить Т-ДНК реплицируется, в результате чего образуется однонитчатая ДНК (ssDNA - single-stranded DNA), получившая название Т-нить. Точно известно, что именно такая однонитчатая Т-ДНК переносится в растительную клетку. После вырезания Т-ДНК VirD2 остается ковалентно связанным с ее 5'-концом и действует как направляющий протеин при ее движении из бактериальной клетки в растительную, а также предохраняет 5'-конец от действия эндонуклеаз.
Перенос Т-ДНК через мембраны бактериальной клетки происходит при участии 11 протеинов-продуктов генов virB (VirB 1-11), а также VirD4. Они образуют транспортный комплекс секреторной системы IV типа (T4SS). Секреторная система IV типа имеется у многих грамотрицательных бактерий, она служит для конъюгативного переноса плазмид между бактериями, а также для перемещения факторов вирулентности от патогенов к клеткам хозяина во время инфекции. Названные протеины формируют так называемый трансмембранный поровый комплекс, который устанавливает связь между наружной и внутренней мембранами бактериальной клетки, и через который происходит перемещение Т-комплекса. Протеины VirB4 и VirB11 являются связанными с мембранами АТФазами, что говорит о том, что процесс переноса Т-комплекса активен и осуществляется с затратой энергии. Каким образом Т-ДНК далее проникает в растительную клетку, точно не установлено. Предполагается, что в этом процессе могут участвовать бактериальные пили, которые могут выполнять роль полой иглы: протыкать оболочку и мембрану растительной клетки, а Т-ДНК по их каналу попадает в цитоплазму. Внутрь растительной клетки поступают и другие макромолекулы, в частности протеины VirE2, VirE3, VirF, VirD5, необходимые для доставки Т-ДНК к ядру хозяйской клетки и включения в ее геном.
В растительной клетке VirE2 плотно покрывает Т-нить по всей ее длине (образуется зрелый Т-комплекс, имеющий спиралеобразную структуру), предохраняя ее от деградации эндонуклеазами. Этот комплекс из-за относительно больших размеров не может попасть в ядро растительной клетки с помощью диффузии. Его доставка осуществляется с помощью бактериальных протеинов, выполняющих роль эффекторов вирулентности, которые, взаимодействуя со специфическими белками и системами растительной клетки, способствуют перемещению Т-комплекса к ядру, хроматину и встраиванию Т-ДНК в геном растительной клетки. Предполагается, что перемещение Т-комплекса к ядру происходит вдоль микротрубочек с помощью двигательных протеинов типа DLC3 (англ. dynein-like protein). Этот процесс, а также преодоление пор ядра (их диаметр 9 нм меньше диаметра Т-комплекса 15 нм) является активным, с затратой энергии. Установлено, что VirE2 взаимодействует со специфическим растительным белком защитного ответа растения на инфекцию VIP1. Его фосфорилирование в случае инфекции сопровождается переносом в ядро, где он активирует связанный с патогенезом (англ. patogénesis related) ген PR1, а также другие гены ответа на стресс (т. е. является транскрипционным фактором). Благодаря тому, что VIP1 в силу своей функциональной ответственности взаимодействует с хроматином растительной клетки, в область хроматина доставляется и связанный с ним Т-комплекс. Для видов растений, у которых вырабатывается небольшое количество VIP1, его функцию в агробактериальной трансформации может выполнять бактериальный белок VirE3. Проникновению Т-комплекса в ядро также способствует наличие у него белка VirD2. VirD2 в случае его синтеза в эукариотических клетках является ядерным белком, который непосредственно взаимодействует с импортином-альфа - элементом ядерного транспорта.
Интеграция Т-ДНК в геном растения завершает процесс ее переноса. Анализ последовательностей ДНК вокруг места вставки показал, что этот процесс не является сайт-специфичным, и не требует высокой степени гомологии (негомологичная рекомбинация). Встраивание Т-ДНК не происходит в строго определенные места (сайты) растительного генома, а в значительной степени случайно. Тем не менее отмечено, что вставки чаще оказываются в области транскрипционно активного хроматина, в частности вблизи теломер хромосом растений. Обычно встраивается одна или несколько копий Т-ДНК в одном или нескольких местах растительного генома. Нередко интеграция Т-ДНК в геном не происходит вообще.
Т-ДНК встраивается в основном в местах двунитевых разрывов с участием ферментов ДНК-репарации растительной клетки. Перед встраиванием Т-ДНК освобождается от защитного покрытия VirE2 (в этом процессе принимает участие убиквитин-протеосомная система (UPS - ubiquitin-proteasome system) растительной клетки, в состав которой включается бактериальный F-box протеин VirF, а протеин VirD5 предохраняет ее от деградации) и происходит дупликация комплементарной нити Т-ДНК (т. е. она превращается в двунитевую ДНК). При этом возможно соединение двух и более Т-ДНК, причем разными концами (B. Lacroix и др., 2011).
Таким образом, агробактерии для осуществления трансформации не только приспособили свою секреторную систему (для переноса Т-ДНК в растительную клетку), но также используют систему защиты растительной клетки от инфекции (для перемещения Т-ДНК в ядро и контакта с хроматином) и систему репарации ее ДНК (для интеграции ДНК в геном).
В случае если встраивание Т-ДНК прошло без осложнений, гены, расположенные на ней, стабильно экспрессируются. Так называемые onc-гены (онкогены) определяют сверхпродукцию фитогормонов ауксина индолилуксусной кислоты и цитокининов, что вызывает неконтролируемый рост и деление трансформированных клеток. В результате образуется корончатый галл. Как отмечалось выше (раздел 3.4), клетки корончатого галла способны расти в культуре in vitro на питательной среде без фитогормонов, поскольку они в состоянии обеспечить ими себя самостоятельно. Активность другой группы генов, расположенных на Т-ДНК, связана с образованием опухолеспецифичных метаболитов - опинов (октопинов, нопалинов, агропинов).
Как видно из представленного описания агробактериальной трансформации, ни онкогены, ни гены, кодирующие образование опинов, в самом процессе переноса Т-ДНК из агробактерии в клетку растений никакой активной роли не выполняют. В этом процессе существенную роль играет лишь крайне незначительная часть Т-ДНК, представленная левым и правым краями из 25 п. н. Если удалить ту часть Т-ДНК, которая расположена между ее краями, и встроить туда нужные гены с соответствующими регуляторными элементами, то они могут быть перенесены и встроены в геном растения с помощью агробактерии точно так же, как она это делает со своими собственными генами. Именно на этом принципе построено использование агробактериальной трансформации в генетической инженерии растений.
Cis-векторы, или коинтегративные, получают путем вставки нужных генов в Т-область Ti-плазмиды Agrobacterium с помощью гомологичной рекомбинации. TVans-векторы, или бинарные, не имеют гомологии с Т-ДНК. Конструируют их исключительно методами технологии рекомбинантных ДНК, описанными выше (встраивание нужных фрагментов ДНК между левым и правым краями осуществляют с помощью полилинкеров). Они обязательно содержат сайт начала репликации (ori) от плазмиды с широким кругом хозяев, либо содержат ori-сайты как Agrobacterium, так и E. coli, благодаря чему способны автономно реплицироваться в этих микроорганизмах (рис. 9.12).
Рис. 9.12. Пример плазмидного бинарного вектора (Вектор pMON10117 использован при создании томатов с удлиненным сроком созревания благодаря встраиванию гена фермента АСС-деаминазы, играющего важную роль в биосинтезе этилена. По внешнему периметру плазмиды отмечены сайты рестрикции соответствующих рестриктаз, по внутреннему периметру - последовательность генетических элементов, содержащихся на плазмиде. Ori-322 и ori-V - сайты начала репликации плазмиды соответственно в E. coli и A. tumefaciens. В геном трансгенных растений включена часть плазмиды, ограниченная левым и правым краями (left border, right border), - T-ДНК.) (по APHIS-USDA, 1994)
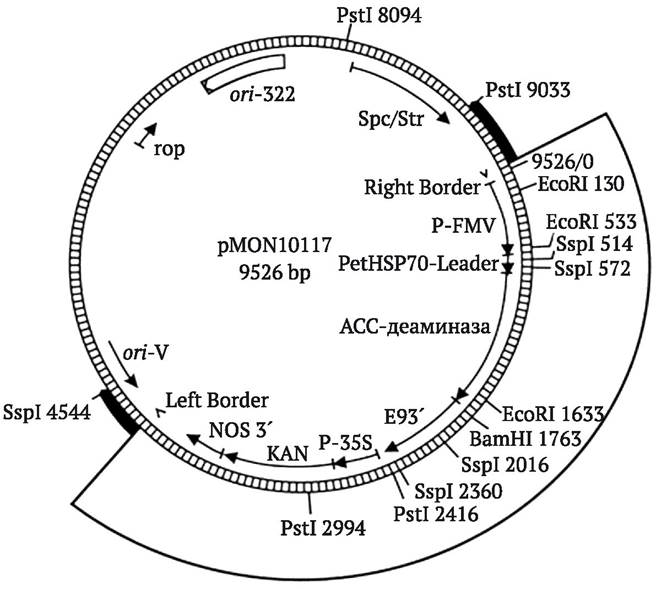
При создании современных трансгенных сортов растений в основном используют бинарные векторные системы. После клонирования в E. coli, изучения и отбора нужных конструкций генов, отобранные бинарные вектора переносят в специальные штаммы Agrobacterium. Их создают на основе высоко вирулентных штаммов Agrobacterium (как правило, A. tumefaciens). Характерной особенностью этих штаммов является то, что они имеют так называемую разоруженную (англ. disarmed), или хэлперную (англ. helper), vir-плазмиду. Последняя представляет собой Ti-плазмиду, которая содержит интактную vir-область, но у которой полностью удалена Т-область. Привнесенный в Agrobacterium бинарный вектор способен в ней автономно реплицироваться благодаря наличию ori-сайта от плазмиды с широким кругом хозяев. Благодаря же наличию разоруженной vir-плазмиды его гены, расположенные в Т-области, могут успешно переноситься и встраиваться в геном клеток растений.
Как видим, при разработке бинарных векторных систем использована такая важнейшая особенность механизма агробактериальной трансформации, согласно которой vir-область Ti-плазмиды лишь обеспечивает перенос Т-ДНК из бактерий в растения, но сама при этом в растения не попадает. С другой стороны, в процессе переноса Т-ДНК существенным является лишь присутствие очень небольшой ее части по 25 п. н. левого и правого края. Остальная часть Т-области, в том числе все онкогены, и гены, кодирующие образование опинов, для процесса переноса Т-ДНК несущественны. Они выполняют в нем пассивную роль и могут быть безболезненно заменены любыми другими генами.
В бинарных векторных системах, в отличие от естественных штаммов Agrobacterium, основные участники процесса агробактериальной трансформации: vir- и chv-гены, которые обеспечивают перенос Т-ДНК, и переносимая Т-ДНК (точнее ее левый и правый край со встроенными между ними нужными генами), разделены физически. Chv-гены (хромосомные вирулентные гены) расположены на бактериальной хромосоме, а vir-гены и Т-ДНК расположены на разных плазмидах. Это очень удобно для генетических манипуляций с бинарным вектором, существенно увеличивает его емкость.
В целях осуществления агробактериальной трансформации растений применяют два основных подхода: кокультивирование суспензии агробактерий с различными эксплантатами (листовыми дисками, эмбриоидами, стеблевыми черенками и др.) или суспензией клеток и трансформацию in planta. Кокультивирование проводят в течение определенного срока (от нескольких часов до нескольких суток), затем эксплантаты или клеточную суспензию переносят на питательную среду, в которую добавляют антибиотики, необходимые для подавления бактерий. После освобождения от бактериальной инфекции проводят отбор трансформированных клеток на селективной среде (если это предусмотрено протоколом), затем из каллюса регенерируют растения. Кокультивирование - основной метод получения трансгенных растений: большинство из них получено с его помощью.
Трансформация in planta предполагает инокуляцию суспензией агробактерий семян или различных органов растений. Семена с подвергнутых воздействию агробактерий растений (или выращенных из семян, обработанных агробактериями) проращивают на селективной среде и отбирают трансформанты. Простейший способ трансформации in planta -замачивание семян в суспензии агробактерий. Для повышения эффективности процесса семена подвергают воздействию ультразвука, делают на них насечки скальпелем, используют вакуумную инфильтрацию, применяют пункцию агробактерий с помощью шприца и тонкой иглы. Также инокулируют агробактериями стеблевые меристемы, цветочные почки. Хорошие результаты были получены при проведении трансформации in planta во время процесса оплодотворения (агробактериями инокулируют пестики и/или пыльцу). Считается, что пыльцевые трубки во время их роста в пестиках - наиболее чувствительные мишени для агробактерий.
Безусловно, агробактериальная трансформация - наиболее эффективная технология введения трансгенов в клетки растений. Однако она имеет ограничения, связанные с кругом хозяев агробактерий. Многие двудольные и голосеменные и практически все однодольные растения устойчивы к агробактериям и никогда не образуют опухолей. Для многих однодольных растений, особенно злаковых, характерна пониженная регенерационная способность из каллюса. Благодаря оптимизации технологии в лабораторных условиях стала возможной агробактериальная трансформация широкого круга высших растений, а также грибов и даже животных клеток. В частности, разработаны протоколы агробактериальной трансформации риса (Y. Hiei и др., 1994), кукурузы (Y. Ishida и др., 1996), пшеницы (M. Cheng и др., 1997) и других злаковых культур.
Расшифровка основных механизмов процесса, изучение факторов, которые могут оказывать на него влияние, позволили существенно повысить эффективность агробактериальной трансформации. Были отобраны высоковирулентные штаммы агробактерий, на основе которых созданы эффективные бинарные векторы. Появились так называемые супербинарные векторы, которые обладают повышенной вирулентностью благодаря добавке дополнительной хэлперной плазмиды с отдельными vir-генами. Разработаны методы нанесения на эксплантаты большого количества очень мелких повреждений с помощью ультразвука или бомбардировкой микрочастицами, что обеспечивает увеличение количества мест контакта агробактерий с растительными клетками. Добавка в питательную среду для суспензии агробактерий ацетосирингона и других веществ, активирующих vir-гены, сделала рутинным применение метода для культур, которые в природе устойчивы к агробактериям. Хорошие результаты были получены при использовании антиоксидантов цистеина, нитрата серебра, аскорбиновой кислоты и других, уменьшающих неблагоприятные для трансформации эффекты защитных реакций растений на инфекцию агробактериями. Существенно повысить эффективность трансформации в каждом конкретном случае позволяет оптимизация таких факторов, как сочетание генотипов агробактерии и растения, температура, состав среды для инокуляции, продолжительность кокультивирования, питательные среды и условия культивирования трансформированных клеток, регенерации растений.
9.5. Экспрессия трансгенов
Экспрессия трансгенов у высших растений может происходить без вставки (транзиентная экспрессия) и после вставки в геном растительной клетки (стабильная экспрессия). Транзиентная экспрессия имеет место непосредственно после проведения агробактериальной трансформации: в течение 2-4 дней после нее наблюдают пик активности трансгенов, которая постепенно падает (уменьшается как число экспрессирующих трансгены клеток, так и уровень экспрессии на клетку). Явление транзиентной экспрессии нашло применение в молекулярно-генетических исследованиях для быстрой доставки в клетки определенных белковых продуктов. В частности, наиболее точный метод детекции в растениях генов вертикальной устойчивости к болезням и вредителям (R-генов) включает в себя агробактериальную инфильтрацию с определенными генами авирулентности патогена. Экспрессия в растениях этих генов приводит к образованию эффекторов, которые, взаимодействуя с продуктами комплементарных R-генов, вызывают характерную реакцию сверхчувствительности (программируемой клеточной смерти), наблюдаемую визуально через короткое время (V. Vleeshouwers и др., 2011).
Стабильная экспрессия трансгенов появляется через 10-14 дней после трансформации. Она может сохраняться на высоком уровне в течение длительного времени и передаваться потомству. Для целей селекции трансгенных сортов растений стабильное проявление привнесенного признака является одним из наиболее важных. Добиться его во многих случаях весьма сложно. Многие из полученных трансформантов несмотря на наличие подтвержденного факта трансгенной вставки этим признаком не обладают (отсутствует экспрессия трансгенов), или обладают в недостаточной мере (слабая экспрессия), или постепенно его теряют, он не сохраняется при размножении (замолкание трансгенов или утрата вставки).
Существует достаточно много факторов, оказывающих влияние на стабильность экспрессии трансгенов. Прежде всего это естественные механизмы защитной реакции растения на внедрение чужеродного генетического материала, которые могут проявляться в гибели трансформированных клеток, внутригеномных перестройках, активации РНК-индуцированного комплекса замолкания генов, метилировании привнесенных участков ДНК. Учитывать их и каким-либо образом оказывать на них влияние сложно.
Вместе с тем имеется ряд факторов, существенно снижающих степень экспрессии трансгенов и непосредственно связанных с процессом генетической модификации, благодаря чему их можно минимизировать. В частности, известно, что экспрессия трансгенов снижается при вставке очень больших фрагментов ДНК, в случае встраивания нескольких копий вставки, при значительных перестройках встроенной ДНК (делециях, появлении инвертированных повторов), что сравнительно часто наблюдается при использовании таких методов переноса трансгенов в геном растений, как биолистика. В связи с этим в селекции трансгенных сортов стараются использовать насколько это возможно наиболее эффективный и предсказуемый метод агробактериальной трансформации. Большое внимание уделяют оптимизации состава трансгенной вставки. В частности, предпочтение отдается модифицированным генам и регуляторным элементам, что обеспечивает более высокую стабильность и высокий уровень экспрессии трансгенов в растениях. Например, вместо полноразмерных нативных генов используют их укороченные версии, кодирующие только активную часть протеина.
С помощью технологии рекомбинантных ДНК удается существенно повысить эффективность существующих промоторов за счет присоединения к ним различных регуляторных элементов, усиливающих экспрессию стоящих под ними генов. В качестве примера можно привести химерную регуляторную последовательность, использованную в толерантной к гербициду глифосату трансгенной сое MON 89788 для экспрессии гена фермента EPSPS, которая включает в себя: промотор гена tsfl фактора элонгации EF1 Arabidopsis thaliana, энхансер промотора FVM (вируса мозаики норичника), лидерную последовательность и интрон гена tsfl A. thaliana, последовательность хлоропластного транзитного пептида. Для усиления экспрессии целевого признака также используют усиленные промоторы, составленные из нескольких копий промоторов.
Промоторы целевого и селективного гена в трансгенных конструкциях могут взаимодействовать между собой, если они близко расположены. Это может привести к непредсказуемой и нестабильной экспрессии соответствующих генов. То же может случиться, если вставка трансгена произойдет вблизи промоторной области какого-либо гена растений. В связи с этим при создании трансгенных конструкций рекомендуют ориентировать целевой и селективный ген таким образом, чтобы их промоторы были максимально удалены друг от друга, или между ними помещают какую-либо нейтральную последовательность - спэйсер. Также для этой цели можно использовать для селективных генов такой промотор, как tCUP (конститутивный криптический промотор табака), который не взаимодействует с другими промоторами.
Экспрессия трансгенов в значительной степени зависит от их первичной структуры. Как отмечалось выше (раздел 9.3.1), в генетической инженерии в качестве целевых часто используют прокариотические гены, которые могут существенно отличаться по нуклеотидному составу от эукариотических
генов. В ДНК бактерий относительное содержание A + T, как правило, выше, чем у растений. В то же время высокое содержание А + Т характерно для сайтов полиаденилирования и других терминальных последовательностей, сайтов сплайсинга, сигналов деградации мРНК эукариот. По частоте использования кодонов бактериальные гены также значительно отличаются от генов растений. Так, в растениях редко встречаются сочетания динуклеотидов CG и TA в позициях «2» и «3» кодонов, а также нуклеотид G в позиции «3» кодонов, кодирующих треонин, пролин, аланин и серин. Проанализировав сиквенс предполагаемых целевых генов с помощью специальных компьютерных программ, можно выявить перечисленные нежелательные в растениях кодоны. После этого производят генно-инженерными методами замену в них нуклеотидов, получая синонимичные кодоны (т. е. кодоны одних и тех же аминокислот), характерные для растительных генов. С помощью этой методики удалось, в частности, существенно повысить экспрессию в растениях генов инсектицидных протеинов Bacillus thuringiensis (M. Adang и др., 1993).
Важным фактором, существенно влияющим на активность привнесенных генов, является случайный характер вставки трансгенов в геном растений. В зависимости от места встраивания степень экспрессии трансгенов может различаться в тысячи раз: от полного молчания до активной стабильной экспрессии. Это явление получило название «эффект положения» (gene position effect) трансгена. Чтобы минимизировать эффект положения вставки, получают большое количество первичных трансформантов, среди которых в дальнейшем отбирают генотипы с требуемым уровнем экспрессии трансгенов. Решить эту проблему можно и с помощью генно-инженерных подходов: путем введения в трансгенные конструкции так называемых MARs (matrix attached regions) - специальных последовательностей, которые предположительно обеспечивают повышенную экспрессию расположенных рядом с ними генов за счет изменения конфигурации петель ДНК относительно ядерного матрикса (гены, расположенные на малых петлях ДНК, экспрессируются сильнее, чем те, что располагаются на больших петлях) (L. Mlynarova, J. Nap, 1997). Следует, однако, иметь в виду, что последовательности MARs, так же, как и селективные гены с их регуляторными элементами, не являются в трансгенных конструкциях обязательными элементами, необходимыми для проявления привнесенного признака, что делает их использование нежелательным.
9.6. Отбор и молекулярно-генетичекий анализ трансгенных растений
Для получения трансгенных растений клеточные клоны, отобранные на селективной питательной среде, переносят на регенерационную среду. В целях повышения эффективности отбора трансформантов добавляют селективный агент и в регенерационную среду, а также в питательные среды, применяемые для укоренения растений-регенерантов, их клонального размножения in vitro. Среди высаженных в грунт растений-регенерантов или растений, отобранных на селективных средах после трансформации in planta, выделяют экземпляры с привнесенным признаком (если это можно сделать визуально), выбраковывают те, что имеют выраженные отклонения в росте и развитии (если это не было целью генетической модификации). Далее проводят селекционную оценку трансформантов. В качестве кандидата на новый сорт могут претендовать только трансгенные линии с максимальным проявлением нового признака и минимальным отличием от исходного сорта по остальным признакам.
Предположительно трансгенные растения подлежат молекулярно-генетическому анализу, который позволяет подтвердить факт трансгенной вставки и экспрессии встроенных генов, получить информацию о характере генетической модификации, ее стабильности, об отсутствии непреднамеренных изменений при ее переносе в геном растения.
Для доказательства наличия трансгенной вставки в геноме анализируемых растений чаще всего применяют ПЦР-детекцию в их ДНК специфических последовательностей целевого гена или других элементов, представленных в использованной генетической конструкции. Чтобы исключить ложноположительные результаты, обусловленные наличием в сосудистой системе растений выживших агробактерий, проводят детекцию специфических для них генов, отсутствующих в трансгенной вставке, например, гена virE2 (ПЦР-продукта этого гена не должно быть). Наличие трансгенной вставки можно также подтвердить с помощью блоттинга по Саузерну, используя в качестве зонда фрагмент ДНК целевого гена. Этим методом определяют и количество копий вставки (по числу и размерам способных гибридизоваться с зондом фрагментов ДНК трансгенных линий, подвергнутых рестрикции, при которой один сайт рестрикции располагается в пределах перенесенной последовательности ДНК, а второй сайт - за ее пределами в произвольном участке генома растений). С помощью анализа расщепления по целевому и/или селективному гену в потомстве трансгенной линии от самоопыления или от скрещивания с исходным сортом определяют стабильность и характер наследования встроенных генов при половом размножении. Наличие селективного гена у сеянцев выявляют, проращивая семена на селективной среде, или с помощью ПЦР-детекции, целевого гена - с помощью ПЦР-детекции.
Индикатором экспрессии в трансгенном растении встроенных генов является образование соответствующих мРНК. Для их выявления используют метод нозерн-блоттинга (аналогичный Саузерн-блоттингу, отличающийся тем, что изучаемым субстратом служит мРНК), а также ОТ-ПЦР. В последнем случае выделяют из анализируемых растений РНК, затем получают кДНК, в которой выявляют последовательности целевого и других встроенных генов с помощью специфичных для них праймеров. Секвенирование кДНК встроенных генов дает возможность определить последовательность нуклеотидов и с помощью специальной компьютерной программы последовательность аминокислот их экспрессируемых продуктов. Эта информация имеет важное значение для определения токсического и аллергенного потенциала этих продуктов при оценке биобезопасности полученных ГМО (путем сравнения их строения с данными по аминокислотному составу известных токсинов и аллергенов по международным базам данных).
Экспрессию встроенных генов оценивают также по наличию у них соответствующих белковых продуктов. Для их выявления используют вестерн- блоттинг (зонды - меченые специфичные к определяемому белку антитела), иммуноферментный анализ (ELISA), другие аналитические методы обнаружения определенных протеинов. Некоторые из этих методов, например, ELISA, обеспечивают возможность количественного определения содержания белка в различных органах и тканях. Об экспрессии трансгенов у ГМ-растений во многих случаях можно судить визуально по степени проявления у них привнесенных признаков, например, удлинении сроков созревания, инсектицидной активности и др.
Основные этапы отбора, молекулярно-генетического анализа и первичной селекционной оценки трансгенных линий представлены на рис. 9.13. Эти результаты являются обязательным и одним из наиболее важных разделов досье ГМО, которое готовят в соответствии с законодательством в области биобезопасности (см. гл. 11). Также в досье среди прочей информации должно быть представлено описание аналитического метода, позволяющего идентифицировать данную трансгенную линию, в том числе в переработанных продуктах питания, сырье и кормах.
Рис. 9.13. Основные этапы отбора, молекулярно-генетического анализа и первичной селекционной оценки трансгенных растений
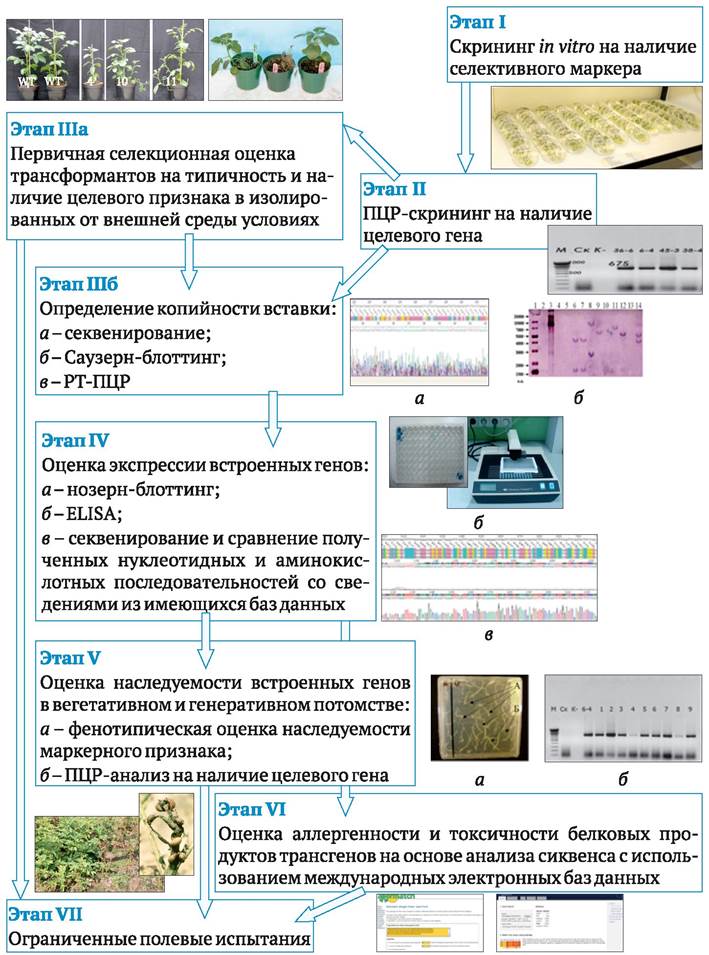
9.7. Детекция ГМ-копонентов в продуктах питания и кормах
Учитывая право потребителей на получение информации о продуктах питания, в том числе о том, что продукт содержит генетически модифицированные составляющие (далее - ГМС), во многих странах принята система соответствующей маркировки. В частности, в Беларуси норма о маркировке ГМО закреплена в Законе «О качестве и безопасности продовольственного сырья и пищевых продуктов для жизни и здоровья человека» и Законе «О защите прав потребителей». В соответствии с законодательством при гигиенической регистрации ряд видов продовольственного сырья и продуктов питания (определенных утвержденным правительством перечнем), которые могут вырабатываться из трансгенных растений, подлежит обязательному лабораторному исследованию на наличие ГМС. Для проведения таких анализов создана сеть аккредитованных лабораторий.
Присутствие ГМС в анализируемых образцах можно обнаружить путем выявления в них белковых продуктов, определенных трансгенов или повторностей ДНК, характерных для ГМО. Первый из названных подходов, основанный на применении ELISA, на практике практически не используется из-за ряда ограничений. Разработаны стандартные методики лишь для ограниченного числа трансгенных белков (CP4 EPSPS, некоторых инсектицидных протеинов). Метод не позволяет идентифицировать конкретные трансгенные линии. Многие белки в процессе переработки сырья разрушаются. Чувствительность этого метода ниже, чем у методов, основанных на анализе ДНК.
Для детекции ГМС в растительном сырье, пищевых продуктах и кормах в основном используют различные методы ПЦР-анализа. Разработаны и приняты следующие стандартые методики детекции ГМС. Наиболее простой и дешевый метод включает выделение ДНК из анализируемого образца, проведение ПЦР со специфическими праймерами и электрофореза продуктов амплификации. Чувствительность этого метода - около 0,1 % ГМС (рис. 9.14). Второй метод основан на детекции ГМС с помощью иммобилизованных на биочипах специфических ДНК-зондов. Его чувствительность приблизительно такая же, как и у первого из названных методов. Третий метод, ПЦР в реальном времени, наиболее автоматизирован и обладает наиболее высокой чувствительностью (около 0,01 % ГМС). Он обеспечивает не только детекцию ГМС, но и их количественное определение. Его недостатком является высокая стоимость оборудования и реактивов.
В зависимости от особенностей национального законодательства по маркировке ГМО применяют два основных направления детекции ГМС: 1) скрининговые исследования; 2) идентификация отдельных трансгенных линий (в англоязычной литературе используется термин «transgenic event» -трансгенное событие, которое характеризуется генетической конструкцией вставки, видом растения, местом и количеством копий вставки) и количественное определение их содержания в образце. При проведении скрининговых исследований рекомендуется использовать праймеры к генетическим элементам, широко применяемым при создании ГМО: к 35S промотору, к терминальной последовательности NOS, маркерному гену nptII. В случае применения в скрининговых исследованиях биочипов помимо названных генетических элементов также выявляют репортерный ген gus и промотор гена ocs (МУК 4.2 230407, приложение к постановлению Главного государственного санитарного врача Российской Федерации от 30 ноября 2007 г. № 80).
Рис. 9.14. Результаты исследования стандартным методом (ПЦР + электрофорез) референсных материалов сои GTS40-3-2 и кукурузы МОN810, ВТ176 с различным содержанием ГМС на предмет выявления повторности ДНК, специфичной для 35S промотора вируса мозаики цветной капусты (нижняя полоса на электрофореграммах), и видоспецифических генов сои и кукурузы (верхняя полоса): 1 - отрицательный контроль (дистиллированная вода); 2 - ERМ 413b (кукуруза, содержащая 0,1 % МОN810); 3 - ERМ 411b (кукуруза, содержащая 0,1 % ВТ176); 4 - ERМ 413а (кукуруза, содержащая менее 0,02 % МОN810); 5 - ERМ410b (соя, содержащая 0,1 % GТS40-3-2); 6 - ERМ413а (соя, содержащая менее 0,03 % GТS40-3-2); 7 - соя, положительный контроль (1,0 % GТS40-3-2); 8 - кукуруза, положительный контроль (1,0 % МОN810). Метод позволяет выявлять ГМС в концентрации 0,1 % и выше (дорожки 2, 3, 5, 7 и 8)
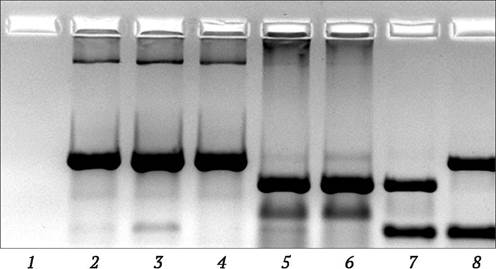
Для идентификации отдельных трансгенных линий используют специфические для них праймеры, которые обеспечивают при проведении ПЦР амплификацию фрагментов ДНК, включающих в себя ДНК вставки и прилегающие к вставке последовательности геномной ДНК растения. При проведении таких исследований в целях сужения поля поиска могут применяться праймеры, позволяющие детектировать определенные целевые гены или генетические конструкции (например, амплификация ДНК, расположенной между определенным целевым геном и определенным промотором). Для проверки качества выделяемых из исследуемых образцов препаратов ДНК применяются методы идентификации последовательностей ДНК, характерных для растений, например, гена лектина сои или гена зеина кукурузы.
При проведении скрининговых исследований анализируют большое количество образцов каких-либо продуктов, которые с определенной вероятностью могут содержать ГМС. Такая система принята, например, в Беларуси, где подлежат обязательному анализу на ГМС продукты из сои и кукурузы, включенные в утвержденный перечень (приложение к постановлению Министерства здравоохранения Республики Беларусь и Комитета по стандартизации, метрологии и сертификации при Совете Министров Республики Беларусь от 08.06.2005 № 12/26):
1. Соя.
2. Соевые бобы.
3. Соевые проростки.
4. Концентрат соевого белка и его текстурированные формы.
5. Изолят соевого белка.
6. Гидролизат соевого белка.
7. Соевая мука и ее текстурированные формы.
8. Заменитель молока (соевое молоко).
9. Заменитель сухого молока (сухое соевое молоко).
10. Консервированная соя.
11. Вареные и жареные соевые бобы.
12. Жареная соевая мука.
13. Продукты, полученные из/или с использованием изолята соевого белка, концентрата соевого белка, гидролизата соевого белка, соевой муки, сухого соевого молока.
14. Ферментированные соевые продукты.
15. Соевая паста и продукты из нее.
16. Соевый соус.
17. Продукты, полученные из/или с использованием соевого молока (тофу, сквашенные напитки, мороженое, майонез и др.).
18. Кукуруза.
19. Кукуруза для непосредственного употребления в пищу (мука, крупа и др.).
20. Кукуруза замороженная и консервированная.
21. Попкорн.
22. Кукурузные чипсы.
23. Мука смешанная, содержащая кукурузную муку.
24. Пищевые добавки, содержащие продукты из сои и (или) кукурузы.
25. Детское питание, полученное с использованием продуктов из сои и (или) кукурузы.
Выбор продуктов сделан на основании анализа посевных площадей, занятых в мире под трансгенными культурами, а также по результатам мониторинговых исследований продуктов, представленных на рынке. Анализ наличия у допущенных к использованию трансгенных линий сои и кукурузы генетических элементов, рекомендуемых для выявления при проведении скрининговых исследований, показал, что 35S промотор вируса мозаики цветной капусты содержат 11 из 17 линий сои и 48 из 54 линий и гибридов кукурузы (по состоянию на ноябрь 2013 г.). Остальные рекомендуемые генетические элементы (терминатор NOS, маркерные гены nptII и ген gus, промотор гена ocs) представлены лишь у небольшого количества линий. Таким образом, методы детекции ГМС, основанные на выявлении в анализируемых образцах последовательностей ДНК 35 S-промотора, позволяют охватить большинство ГМ-продуктов, поступающих на рынок Беларуси. Принимая во внимание, что маркировка ГМС не связана с их биобезопасностью, а проводится исключительно в целях информирования населения, детекция ГМС только по 35S-промотору позволяет сэкономить значительные средства без существенного снижения качества исследований. В отдельных случаях, например, при анализе образцов детского питания, содержащих кукурузу, для детекции трансгенной линии GA-21, у которой отсутствует 35S-промотор, целесообразно использовать праймеры, специфичные для этой линии.
Законодательством Европейского союза и Российской Федерации установлен минимальный порог 0,9 % на присутствие в сырье, продуктах и кормах биологического материала, зарегистрированных трансгенных линий относительно аналогов традиционной селекции (немодифицированных), превышение которого требует обязательной маркировки соответствующих продуктов, сырья и кормов. Введение названного порога объясняют тем, что технически во многих случаях сложно избежать случайного занесения ГМ- примесей в готовую продукцию при ее производстве, транспортировке и торговом обороте. Идентификация трансгенной линии (события) в образце позволяет предупредить помещение на рынок неавторизованных ГМО. Следует также иметь в виду, что корректное количественное определение содержания ГМС возможно только в случае точной идентификации трансгенной линии.
Количественный анализ содержания ГМС проводят с помощью метода ПЦР в реальном времени на специальных термоциклерах, позволяющих учитывать нарастание флюоресценции, происходящее в ходе амплификации определенных детектируемых фрагментов ДНК со специфическими праймерами и мечеными флюоресцентными красителями зондами. В анализируемых образцах с помощью мультиплексной ПЦР (т. е. одновременной ПЦР с несколькими праймерами и зондами) определяют нарастание флюоресценции по детектируемому специфическому для трансгенной линии фрагменту ДНК и фрагменту видоспецифической эндогенной ДНК растения (эндогенный контроль), например, гена лектина (при анализе трансгенных линий сои). Зонды, специфичные к трансгену и эндогенному контролю, метят разными красителями, например, FAM и VIC. На основании полученных кривых (рис. 9.15) для каждого образца определяют величину порогового цикла, при котором кривая флюоресценции переходит в экспоненциальную фазу (Ct) по зонду трансгена и по зонду эндогенного контроля, затем рассчитывают разность между ними (dCt). Определение dCt позволяет нормализовать содержание трансгенной ДНК в каждой пробирке относительно ДНК эндогенного контроля.
Рис. 9.15. График нарастания флюоресценции при амплификации ДНК методом ПЦР в реальном времени образцов трансгенной кукурузы линии MON 810 с разным содержанием ГМС (детекция 35S-промотора, краситель FAM) (красной горизонтальной линией обозначен уровень флюоресценции, соответствующий переходу кривых в экспоненциальную фазу)
Одновременно проводят аналогичный анализ ряда референсных (сертифицированных) материалов определенной трансгенной линии с разным фиксированным содержанием ГМС, например, 0,1, 1,0, 2,0, 5,0 %. Чаще всего используют материалы производства Института референсных материалов Европейского союза - Institute for reference materials andmeasurements (JRC, Directorate-General EC). Референсные материалы готовят путем перемешивания биологического материала (например, муки) немодифицированного аналога (0 % ГМС) с определенным количеством материала трансгенной линии (100 % ГМС). На основании анализа референсных материалов строят калибровочную прямую, используя которую определяют содержание ГМС в анализируемых образцах. Количественное определение ГМС осуществляется с помощью специальных компьютерных программ.
9.8. Получение транспластомных растений
В живой клетке высших организмов ДНК содержится не только в ядре, но и в органеллах: пластидах (хлоропластах, хромопластах, лейкопластах и др.) и митохондриях. При изучении организации геномов пластид и митохондрий выявлено их большое сходство с бактериальными геномами. Это сходство касается структуры кольцевых молекул ДНК, порядка генов и особенностей их организации в полицистроны, параметров белоксинтезирующей системы, чувствительности органелл к антибиотикам и др. Эти данные подтверждают симбиотическую гипотезу возникновения эукариотических клеток, согласно которой предшественниками органелл являются прокариотические организмы.
Метод биолистики позволяет вводить чужеродную ДНК в клетки, в результате чего может происходить трансформация ДНК органелл. Именно разработка метода биолистики позволила получить первые транспластомные растения. В отличие от ядерной ДНК, встраивание трансгенов в которую происходит случайным образом, в случае трансформации пластидной ДНК возможна вставка в определенные сайты с помощью механизма гомологичной рекомбинации. Для этого при конструировании векторов в целях трансформации хлоропластов встраиваемые гены фланкируют последовательностями ДНК (1000-2000 п. н.), гомологичными выбранному сайту генома пластид, в который предполагается трансгенная вставка. В качестве такого сайта используют часть генома хлоропластов, не являющуюся функционально важной, и инсерция в которую не приведет к негативным последствиям для жизнеспособности растения. В первых работах по трансформации хлоропластов в качестве места встраивания трансгенов использовали транскрипционно неактивные области их генома (спэйсеры). В последнее время для этих целей выбирают транскрипционно активные сайты, содержащие инвертированные повторы, встраивание в которые приводит к последующему корректированию числа копий вставки - ее дупликации на противоположном гомологичном сайте с инвертированными повторами. В результате достигается более высокий уровень экспрессии трансгенов. Например, среди часто используемых сайтов инсерции трансгенов у табака область между генами тРНК изолейцина (TrnI) и тРНК аланина (TrnA), а также между геном тРНК валина (TrnV) и опероном rps 12/7 (B. Meyers и др., 2010) (рис. 9.16).
Рис. 9.16. Получение транспластомных растений: а - схема вектора, предназначенного для трансформации пластид, и механизма гомологичной рекомбинации хлоропластов (по B. Meyers и др., 2010) (Встраивание Т-ДНК происходит в специфические участки генома пластид, являющиеся транскрипционно активными сайтами с инвертированными повторами (ИПА и ИПВ). На схеме встраивание трансгена произошло в ИПА между генами тРНК изолейцина (TrnI) и тРНК аланина (TrnA). LSC и SSC - большой и малый однокопийные регионы (англ. large single copy и small single copy) соответственно, RBS - сайт прикрепления к рибосоме (англ. RBS - ribosome binding site), инициирующий трансляцию.); б - экспрессия гена gfpв транспластомном растении свеклы (по F. De Marchi и др., 2009) (На рисунках бI и бIII эпидермис транспластомного растения с содержащими хлоропласты замыкающими клетками устьиц в поле зрения светового микроскопа; на рисунках бII и бIV - то же самое при ультрафиолетовом освещении: видно свечение в хлоропластах продукта трансгена - зеленого флюоресцирующего белка (GFP).)
Полицистронная экспрессия генов, характерная для хлоропластного генома, делает возможным использование трансгенных конструкций только с одним промотором и одним терминатором для экспрессии всех встроенных генов (как единый оперон). Однако при этом каждая открытая рамка считывания требует отдельный сайт прикрепления к рибосоме (англ. RBS - ribosome binding site) для инициации трансляции. Показана высокая эффективность ряда бактериальных и пластидных RBS, имеющих последовательности, подобные последовательности Шайна - Дельгарно, для обеспечения трансляции встроенных генов у транспластомных растений.
Количество копий молекул пластидных ДНК может достигать 10 тыс. (одна клетка может содержать до 100 хлоропластов, в каждом из которых имеется до 100 молекул ДНК). Естественно, добиться одновременного встраивания чужеродных генов во все эти молекулы невозможно. Поэтому одна из основных проблем получения транспластомных форм - достижение гомоплазии трансформированных клеток, т. е. полного исключения у них нетрансформированных пластидных ДНК. В первичных транспластомных клетках последние преобладают: у них имеются лишь единичные молекулы со вставкой привнесенных генов. Для достижения гомоплазии применяется селективная процедура: культивирование «обработанных» рекомбинантными плазмидами тканей на питательной среде с антибиотиком. Делиться на такой среде способны только клетки, в которых имеются трансформированные пластиды. Из каллюсных клеток, полученных на среде с антибиотиком, далее регенерируют растения. Для полного избавления от нетрансформированных пластомов проводят повторные циклы получения каллюса-регенерации растений на среде с высоким содержанием антибиотика.
Трансформация пластид рассматривается как одно из перспективных направлений генетической инженерии растений, поскольку имеет по сравнению с трансформацией ядерной ДНК ряд преимуществ. В силу прокариотической природы ДНК пластид возможно использование механизма гомологичной рекомбинации для сайт-специфического встраивания чужеродной ДНК в их геном подобно тому, как это делают при трансформации прокариот. Благодаря многокопийности ДНК пластид удается добиться очень высокого уровня экспрессии белка-продукта привнесенного гена у транспластомных растений (525 и даже более 40 % транспластомного протеина ко всему клеточному белку; у трансгенных растений лишь 1-2 %). Это делает рассматриваемую модель весьма привлекательной для создания растений-суперпродуцентов ценных веществ. Транспластомные растения по сравнению с трансгенными менее потенциально опасны для окружающей среды, поскольку не способны передавать привнесенные гены путем рассеивания пыльцы (наследование пластомных генов происходит в основном по материнской линии).
Заключение
Генетическая инженерия - это технология получения новых комбинаций генетического материала путем проводимых вне клетки манипуляций с молекулами нуклеиновых кислот и переноса созданных конструкций генов в живой организм, в результате которого достигается их включение и активность в этом организме и у его потомства. Исходя из этого определения, процесс создания генетически модифицированных растений можно разделить на несколько этапов:
✵ выделение и идентификация отдельных генов, которые собираются перенести в растения, а также соответствующих регуляторных элементов;
✵ соединение генов и регуляторных элементов между собой в определенном порядке с помощью технологии рекомбинантных ДНК (создание трансгенных конструкций);
✵ перенос трансгенных конструкций в отдельные живые клетки растения (процесс трансформации, «генетической модификации»), где они могут реплицироваться и передаваться дочерним клеткам, образовавшимся при делении трансформированных клеток;
✵ отбор трансформированных клеток на селективных средах и регенерация растений-трансформантов;
✵ молекулярно-генетический анализ трансформантов в целях подтверждения инсерции трансгенов и их экспрессии, характеристики трансгенной вставки;
✵ отбор трансформантов с оптимальным и стабильным проявлением привнесенных трансгенных признаков.
Центральное звено генетической инженерии - технология получения рекомбинантных молекул ДНК. Она позволяет проводить in vitro манипуляции с фрагментами ДНК, выделенными из живых организмов или синтезированными искусственно, вносить определенные изменения в структуру естественных генов и их регуляторных элементов, комбинировать их в произвольной последовательности.
Для получения отдельных фрагментов ДНК применяют ферменты-рестриктазы, способные узнавать и расщеплять специфические последовательности нуклеотидов (сайты узнавания) в молекуле ДНК. Для генной инженерии наибольшее значение имело выделение рестриктаз, которые дают фрагменты с так называемыми «липкими» (комплементарными) концами. Фермент концевая трансфераза катализирует присоединение нуклеотидов к 3'-концам цепей ДНК. Благодаря этому из «тупого» конца ДНК можно сделать «липкий».
Если объединить в одной пробирке фрагменты ДНК любого происхождения, полученные с помощью одной и той же рестриктазы, дающей «липкие» концы, то эти фрагменты соединятся между собой. Для воссоединения фосфодиэфирных связей между концами сшиваемых фрагментов ДНК применяют фермент ДНК-лигазу. В генно-инженерных работах для произвольного соединения фрагментов ДНК часто используют линкеры - искусственные сайты рестрикции. Линкеры пришивают к фрагменту ДНК, а затем расщепляют их рестриктазой. В результате этот фрагмент приобретает «липкий» конец нужной конфигурации. Линкеры, имеющие несколько сайтов узнавания ре- стриктазами, называются полилинкерами.
Одним из основных элементов технологии рекомбинантных ДНК является клонирование генов. Для клонирования фрагмента ДНК его встраивают в плазмиды (векторы клонирования), полученные рекомбинантные плазмиды далее вводят в бактериальные клетки (обычно это Е. coli), где они могут реплицироваться. Таким образом можно получать многочисленные копии определенных фрагментов ДНК, нарабатывать достаточные объемы для последующего изучения и возможных манипуляций с ними. Помимо бактериальных плазмид для целей клонирования генов используют и другие векторные системы: фаги, плазмиды-космиды, а также так называемые искусственные хромосомы на основе фага Р1, F-плазмиды Е. coli (BAC - бактериальная искусственная хромосома), дрожжей (YAC-дрожжевая искусственная хромосома). В качестве клеток-хозяев для клонирования генов помимо Е. coli используют также Bacillus subtilis, дрожжи Saccharomyces cerevisiae и др.
Возможны два основных подхода в выделении гена: либо его синтезируют, либо выбирают среди рестрикционных фрагментов геномной ДНК тот из них, который содержит нужный ген. Для многих генов определена последовательность нуклеотидов. Эта бесценная информация хранится в международных базах данных и доступна с помощью Интернета для любого исследователя. Современные ДНК-синтезаторы способны присоединять один нуклеотид к цепочке ДНК в течение нескольких минут, формируя фрагменты ДНК длиной более ста пар нуклеотидов. Соединяя отдельные фрагменты ДНК, можно получить полноразмерный ген.
Второй способ искусственного синтеза гена основан на использовании метода полимеразной цепной реакции (ПЦР). Если в международных базах
данных содержится информация о сиквенсе интересующего нас гена какого- либо организма (или близких в систематическом отношении организмов), то с помощью специальных компьютерных программ можно подобрать праймеры, необходимые для амплификации кодирующей последовательности ДНК, используя в качестве исходной матрицы ДНК этого организма. В результате многократного повторения реакций амплификации ДНК образуются многочисленные копии последовательности ДНК, заключенной между праймерами. В реакции амплификации в качестве матрицы может быть использована его мРНК. Соответствующие мРНК выделяют, тщательно очищают и используют для синтеза комплементарной ДНК (кДНК) с помощью фермента обратной транскриптазы, или ревертазы (метод ОТ-ПЦР - ПЦР с обратной транскрипцией).
Для выделения нужного гена из смеси рестрикционных фрагментов используют меченые зонды - фрагменты ДНК, комплементарные искомым генам (целиком или отдельным их частям). Зонды метят с помощью радиоактивного фосфора или хемилюминесцентных меток. В основе метода лежит свойство одноцепочечных молекул ДНК соединяться (гибридизоваться) в случае их комплементарности. Для перевода молекулы ДНК в одноцепочечную форму ее подвергают тепловому воздействию или обработке щелочью (процесс денатурации ДНК). Если в растворе одноцепочечной ДНК присутствует ДНК-зонд, он будет ренатурировать с ДНК, специфически связываясь с комплементарными участками, которые выявляют с помощью метки.
Нужный ген может быть выделен или до, или после процедуры клонирования в E. coli смеси рестрикционных фрагментов геномной ДНК. В первом случае фрагменты ДНК, полученные после обработки рестриктазами, разделяют в соответствии с их размерами с помощью электрофореза в агарозном геле. Поскольку проводить гибридизацию ДНК с зондами в геле невозможно, ДНК после электрофореза переносят на более подходящие для этих целей носители: нитроцеллюлозную или нейлоновую мембраны (блоттинг по Саузерну). Фрагменты ДНК вымываются из геля и закрепляются на нитроцеллюлозе именно в той последовательности, как они расположились после электрофореза. На ней и проводят гибридизацию с меченым ДНК-зондом. Метка позволяет определить местоположение комплементарного фрагмента ДНК на электрофореграмме. При электрофорезе эти фрагменты не разрушаются и их можно выделить (элюировать) из геля, не подвергавшегося блоттингу, в виде биологически активных двухцепочечных молекул.
ДНК-ДНК гибридизацию применяют и для выделения искомых фрагментов ДНК после того, как они были клонированы. Этот подход включает создание и изучение так называемой геномной библиотеки, или клонотеки генов. Клонотека представляет собой колонии бактерий, клетки которых содержат рекомбинантные плазмиды со встроенными в них разными рестрикционными фрагментами ДНК. Эти колонии переносят непосредственно на нитроцеллюлозный фильтр, размещая их в определенном порядке. При подготовке проб к анализу проводят лизис бактериальных клеток и денатурацию содержащейся в них ДНК. Затем осуществляют гибридизацию с меченым зондом и определяют таким образом, в какой колонии клонирован интересующий фрагмент ДНК.
Технология рекомбинантных ДНК позволяет создавать генетические конструкции, содержащие один или несколько целевых генов (генов, кодирующих определенный новый признак, который исследователь собирается придать организму), а также все необходимые регуляторные элементы, обеспечивающие активность (экспрессию) этих генов после их переноса в организмы. В целях упрощения отбора трансформированных клеток в генетические конструкции включают также последовательности, кодирующие селективные или репортерные гены с соответствующими регуляторными элементами.
Основные трансгенные признаки: толерантность к гербицидам, устойчивость к насекомым-вредителям, устойчивость к вирусным болезням, удлинение сроков созревания и способность к длительному хранению, улучшенные качественные характеристики (состава жирных кислот растительного масла, крахмала, пониженное содержание никотина в листьях табака), улучшенные кормовые свойства, система получения гетерозисных гибридов на основе мужской стерильности/восстановления фертильности, устойчивость к засухе, пригодность для производства биотоплива. Постоянно идет поиск новых генов, представляющих практический интерес. Основная масса генов, кодирующих названные признаки, бактериального или вирусного происхождения. Их называют трансгенами. Из-за сомнений общественности относительно безопасности ГМО для здоровья человека и окружающей среды предложено использовать в качестве целевых цис-гены: гены и их собственные регуляторные элементы от растений того же или близкородственного дикого вида, с которым возможна половая гибридизация. В целях изменения активности отдельных генов растений, связанных с контролем качественных характеристик или сроков созревания, применяют перенос генетических конструкций, содержащих кодирующие последовательности этих генов (т. е. генов того же вида растения). Вставка дополнительной копии гена (в смысловой или антисмысловой ориентации относительно промотора) во многих случаях приводит к его замолканию. Использование для целей генетической модификации исключительно собственных генов растений лежит в основе интрагенного подхода при получении ГМО.
Поскольку процесс трансформации затрагивает лишь небольшое количество компетентных клеток, то для их отбора используют селективные маркерные гены. При высеве подвергнутых трансформации клеток на питательную среду, содержащую селективный агент, способностью делиться будут обладать только те клетки, в геном которых произошла вставка рекомбинантной ДНК. В генетической инженерии растений чаще всего в качестве маркерных используют гены устойчивости к антибиотикам и гены устойчивости к гербицидам. Наиболее широкое применение нашел ген nptII неомицинфосфотрансферазы II из E. coli, способный дезактивировать антибиотики канамицин, неомицин, гентамицин. Среди генов устойчивости к гербицидам чаще всего используют гены bar или pat фосфинотрицин N-ацетилтрансферазы - фермента, дезактивирующего фосфинотрицин (глюфозинат аммония). Эти гены особенно эффективны применительно к злаковым растениям, поскольку антибиотик канамицин для однодольных недостаточно токсичен.
Имеется специфическая группа маркерных генов (репортерных), которые способны менять внешний вид трансформированных клеток, благодаря чему их можно визуально отличить от нетрансформированных. Репортерные гены помещают в генетических конструкциях вслед за промотором впереди последовательности, кодирующей целевой ген. Благодаря этому по степени экспрессии репортерного гена (ее оценивают количественно) можно судить и об экспрессии целевого гена. Чаще всего в качестве репортерного гена используют ген gus из E. coli, кодирующий образование фермента β-глюкоронидазы. Экспрессию этого гена оценивают с помощью спектрометрических или гистохимических методов. В тех случаях, когда необходимо проследить динамику экспрессии трансгена в онтогенезе растения, рекомендуют использовать такие гены, как gfp зеленого флюоресцентного протеина (GFP - green fluorescent protein).
Маркерные гены необходимы лишь для отбора трансформированных клеток. В связи с этим разработаны методы получения безмаркерных трансгенных растений. Среди таких методов использование трансгенных конструкций без маркерных генов с отбором трансформантов из большого количества полученных растений-регенерантов, котрансформация (целевой и селективный гены располагаются на разных плазмидах одного штамма агробактерии или на плазмидах разных штаммов) с последующим негативным отбором по селективным генам в расщепляющихся поколениях полученных трансформантов. В целях удаления селективных генов из генома ГМО используют генетические конструкции, содержащие сцепленные с ними последовательности транспозонов, которые обеспечивают их транспозицию и сегрегацию в соматических клетках. Второй подход основан на использовании сайт-специфических рекомбиназ, способных узнавать и удалять из генома специфические последовательности ДНК. Эти последовательности размещают в трансгенной конструкции по краям гена, который планируется позднее удалить.
Для транскрипции встроенного гена обязательно наличие, помимо кодирующей области, также промотора и терминальной последовательности (терминатора). В растительных клетках может функционировать только ген, имеющий эукариотический промотор. Наиболее часто используемые при получении трансгенных растений конститутивные промоторы: CaMV35Sмалой субъединицы вируса мозаики цветной капусты (35S-промотор), промотор гена nos нопалинсинтазы Agrobacterium tumefaciens. Для злаковых культур используют промоторы генов ract1 актина риса и ZmUbi убиквитина кукурузы. Для экспрессии трансгенов в определенных органах растений используют тканеспецифические промоторы: PTA29 из табака (экспрессия трансгенов в пыльниках), промотор гена gbss картофеля (экспрессия в клубнях). Промотор гена rbcS малой субъединицы рибулезобифосфат карбоксилазы-оксигеназы Arabidopsis thaliana - светочувствительный: экспрессия расположенных под ним трансгенов на свету (например, в листьях) в сто и более раз выше, чем в темноте (например, в корнях, клубнях). В трансгенных конструкциях часто используют терминальные последовательности генов nos Agrobacterium tumefaciens и CaMV35S малой субъединицы вируса мозаики цветной капусты, а также различных растительных генов.
Среди методов прямого переноса генов в растительные клетки наибольшее распространение получил метод биологической баллистики (биолистики). Суть метода заключается в том, что на микроскопические частицы вольфрама или золота напыляют рекомбинантные ДНК, разгоняют их до скорости 300-600 м/сек в «генной пушке» и помещают на их пути растительные ткани (меристемы, незрелые зародыши, колеоптили, протокормы, пыльцу) или культуры клеток (каллюс или суспензию клеток). В результате частицы проникают в клетки, а чужеродная ДНК встраивается в ДНК хромосом или органелл. Для прямого переноса генов в растительные клетки также применяют трансформацию протопластов (с использованием электропорации, микроинъекций ДНК).
Наиболее эффективный метод переноса трансгенов в растительные клетки - агробактериальная трансформация. Он основан на использовании уникального природного механизма горизонтального переноса генов плазмид (Т-ДНК) бактерий Agrobactrium tumefaciens и A. rhizogenes в геном растений, в результате экспрессии которых происходит образование опухолей (галлов или «бородатых корней») и биосинтез опинов, утилизируемых бактериями. Агробактерии для осуществления трансформации приспособили свою секреторную систему (для введения Т-ДНК в растительную клетку), систему защиты растительной клетки от инфекции (для перемещения Т-ДНК в ядро клетки хозяина и контакта с хроматином) и систему репарации ее ДНК (для интеграции Т-ДНК в геном).
В результате изучения механизма агробактериальной трансформации было установлено, что ни онкогены, ни гены, кодирующие образование опинов, в процессе переноса Т-ДНК из агробактерии в клетку растений никакой активной роли не выполняют. Существенную роль играет лишь часть Т-ДНК, представленная левым (LB) и правым (RB) краями из 25 п. н. Если с помощью технологии рекомбинантных ДНК встроить в плазмиду агробактерий между LB и RB, вместо Т-ДНК нужные гены с соответствующими регуляторными элементами, то они могут быть перенесены и встроены в геном растения с помощью агробактерии точно так же, как она это делает со своими собственными генами. Именно на этом принципе построено использование агробактериальной трансформации в генетической инженерии растений.
Cis- или коинтегративные вектора получают путем вставки нужных генов в Т-область Ti-плазмиды Agrobacterium с помощью гомологичной рекомбинации. Trans- или бинарные вектора не имеют гомологии с Т-ДНК. Конструируют их исключительно методами технологии рекомбинантных ДНК (в основном с помощью полилинкеров). Они обязательно содержат сайт начала репликации (ori) от плазмиды с широким кругом хозяев, либо содержат ori-сайты как Agrobacterium, так и E. coli, благодаря чему способны автономно реплицироваться в этих микроорганизмах. В бинарных векторных системах, в отличие от естественных штаммов Agrobacterium, основные участники процесса агробактериальной трансформации: vir- и chv-гены, которые обеспечивают перенос Т-ДНК, и переносимая Т-ДНК (точнее ее левый и правый край со встроенными между ними нужными генами), разделены физически. Chv-гены расположены на бактериальной хромосоме, а vir-гены и Т-ДНК - на разных плазмидах. При создании современных трансгенных сортов растений в основном используют бинарные векторные системы.
Экспрессия трансгенов у высших растений может происходить без вставки (транзиентная экспрессия) и после вставки в геном растительной клетки (стабильная экспрессия). Для целей селекции трансгенных сортов растений стабильное проявление привнесенного признака является одним из наиболее важных. Добиться его во многих случаях весьма сложно. Экспрессия трансгенов снижается при вставке очень больших фрагментов ДНК, в случае встраивания нескольких копий вставки, при значительных перестройках встроенной ДНК. В связи с этим в селекции трансгенных сортов стараются использовать, насколько это возможно, наиболее эффективный и предсказуемый метод агробактериальной трансформации. Большое внимание уделяют оптимизации состава трансгенной вставки. В частности, предпочтение отдается модифицированным генам и регуляторным элементам, что обеспечивает более высокую стабильность и высокий уровень экспрессии трансгенов в растениях. Например, вместо полноразмерных нативных генов используют их укороченные версии, кодирующие только активную часть протеина. С помощью технологии рекомбинантных ДНК удается значительно повысить эффективность существующих промоторов за счет присоединения к ним различных регуляторных элементов, усиливающих экспрессию стоящих под ними генов. Экспрессия прокариотических генов может быть существенно повышена за счет модификации их первичной структуры путем синонимических замен определенных нуклеотидов, что позволяет приблизить их по составу кодонов к растительным генам.
Полученные предположительно трансгенные растения подлежат молекулярно-генетическому анализу, который позволяет подтвердить факт трансгенной вставки и экспрессии встроенных генов, иметь информацию о характере генетической модификации, ее стабильности, отсутствии непреднамеренных изменений при ее переносе в геном растения. Далее проводят селекционную оценку трансформантов. В качестве кандидата на новый сорт могут претендовать только трансгенные линии с максимальным проявлением привнесенного признака и минимальным отличием от исходного сорта по остальным признакам.
Учитывая право потребителей на получение информации о продуктах питания, в том числе о том, что продукт содержит генетически модифицированные составляющие, во многих странах принята система соответствующей маркировки. Присутствие ГМС в анализируемых образцах подтверждают путем выявления в них характерных для ГМО повторностей ДНК (исследования продуктов методом ПЦР проводят в аккредитованных лабораториях). При проведении скрининговых исследований рекомендуют использовать праймеры к 35S промотору, к терминальной последовательности NOS, маркерному гену nptII. Для идентификации отдельных трансгенных линий используют специфические для них праймеры, которые обеспечивают при проведении ПЦР амплификацию фрагментов ДНК, включающих ДНК вставки и прилегающие к вставке последовательности геномной ДНК растения.
Контрольные вопросы
1. Дайте определение генетической инженерии.
2. В чем состоит суть технологии рекомбинантных ДНК, назовите ее основные элементы.
3. Какие ферменты используют в технологии рекомбинантных ДНК, для каких целей?
4. Что такое линкеры (полилинкеры), для чего они необходимы при проведении генно-инженерных работ?
5. Каким образом проводят клонирование генов? Какие векторы и организмы используют для клонирования генов?
6. Какие методы используют для искусственного синтеза генов?
7. Как можно выделить определенный ген из смеси рестрикционных фрагментов ДНК?
8. Для какой цели применяют Саузерн-блоттинг?
9. Что такое клонотека генов (геномная библиотека), как можно выделить ген из клонотеки?
10. Какие целевые гены используют для получения трансгенных растений? Чем отличаются цис-гены от трансгенов?
11. Какие гены используют в целях модификации качественных характеристик растений?
12. Почему в трансгенных конструкциях необходимо присутствие селективных генов? Назовите наиболее используемые селективные гены.
13. С какой целью в генно-инженерных исследованиях используют репортерные гены?
14. С помощью каких методов можно получить безмаркерные трансгенные растения? В чем состоит необходимость их получения?
15. Назовите основные промоторы и терминаторы, применяемые в генно-инженерных работах. Чем конститутивные промоторы отличаются от тканеспецифических?
16. Какие методы прямого переноса генов используют для получения трансгенных растений?
17. Опишите процесс переноса Т-ДНК А. tumefaciens в растительную клетку.
18. Что из себя представляют бинарные векторы для переноса генетической информации? Чем они отличаются от коинтегративных?
19. Каким образом повышают эффективность агробактериальной трансформации?
20. Какие методы применяют в целях повышения степени экспрессии трансгенов?
21. Назовите наиболее распространенные праймеры для ПЦР-детекции генетически модифицированных составляющих в продуктах питания.