Молекулярная биология клетки - Том 1 - Албертс Б., Брей Д., Льюис Дж., Рэфф М., Робертс К., Уотсон Дж. 1994
Введение в биологию клетки
Как изучают клетки?
Фракционирование клеточного содержимого
Биохимический анализ часто сопряжен с разрушением тонкой структуры клеток. Однако в настоящее время разработаны методы мягкого фракционирования клеточного содержимого, целью которых является сохранение функции различных клеточных компонентов. Подобно тому как ткань можно разделить на составляющие клетки различных типов, клетки можно разделить на ее функциональные органеллы и макромолекулы. В этом разделе мы сосредоточим внимание на методах, позволяющих проводить очистку органелл и белков. Родственные методы мечения макромолекул радиоизотопами и антителами, равно как и чрезвычайно эффективные методы анализа ДНК и функции генов, обсуждаются в последующих разделах.
4.4.1. С помощью ультрацентрифугирования можно разделять органеллы и макромолекулы [24]
Существует несколько способов разрушения клеток: их можно подвергнуть осмотическому шоку, ультразвуковой вибрации, продавить через маленькое отверстие или измельчить. При этом мембраны клеток (в том числе плазматическая мембрана и мембрана эндоплазматического ретикулума) распадаются на фрагменты, которые сразу же замыкаются, образуя мельчайшие пузырьки. При осторожном применении методов разрушения некоторые органеллы сохраняются в интактном состоянии (ядра, митохондрии, аппарат Гольджи, лизосомы и пероксисомы). Таким образом, суспензия клеток превращается в растворимый экстракт, содержащий довольно грубую суспензию связанных с мембранами частиц, обладающих характерными размерами, зарядом и плотностью. Было показано, что при правильном выборе среды для гомогенизации (а это требует тщательного анализа методом проб и ошибок в отношении каждой из органелл) частицы экстракта сохраняют большую часть биохимических свойств, присущих интактным органеллам в клетке.
![]()
Рис. 4-42. Схематически показано фракционирование субклеточных компонентов из экстрактов клеток путем повторного центрифугирования при постепенно возрастающих скоростях. В общем случае чем меньше по размерам субклеточный компонент, тем более высокая центробежная сила требуется для его осаждения. Обычно на различных этапах центрифугирования требуются следующие условия: низкая скорость - 1000 g - 10 мин, средняя скорость - 20 000 g - 20 мин, высокая скорость - 80000 g - 1 ч, очень высокая скорость - 150000 g - 3 ч.
После того как в начале 40-х годов начали широко использовать препаративную центрифугу, разделение различных компонентов гомогената стало вполне реальным. Экстракты разрушенных клеток фракционируют, подвергая их высокоскоростному центрифугированию (рис. 441). Такая обработка делит клеточные компоненты по их размеру: более крупные частицы при центрифугировании движутся быстрее. Крупные компоненты экстракта, в том числе ядра или неразрушенные клетки, быстро оседают (седиментируют) при относительно низких скоростях и образуют осадок на дне центрифужной пробирки. При более высокой скорости выпадают в осадок митохондрии, а при еще более высоких скоростях и длительных периодах центрифугирования осаждаются мелкие замкнутые пузырьки (микросомы), а затем рибосомы (рис. 4-42). Все эти фракции загрязнены, но если процедуру ресуспендирования осадка и центрифугирования повторить несколько раз, то многие примеси исчезнут.
Центрифугирование является, как правило, первым этапом фракционирования, с его помощью разделяются только значительно отличающиеся по размеру компоненты. Чтобы достигнуть более высокой степени разделения фракций, необходимо гомогенат наслоить тонким слоем поверх солевого раствора. При центрифугировании различные фракции седиментируют с различной скоростью и образуют отдельные полосы, которые можно выделить (рис. 4-43). Во избежание перемешивания осажденных компонентов солевой раствор должен содержать инертный и хорошо растворимый материал (например, сахарозу), плотность которого постепенно увеличивается сверху вниз, формируя градиент плотности.
При седиментации сквозь такие градиенты сахарозы различные компоненты клетки собираются в отдельные полосы, которые можно выделить (см. рис. 4-43). Скорость седиментации каждого из компонентов определяется его размерами и формой и обычно выражается с помощью коэффициента седиментации, обозначаемого S (см. табл. 4-7). Ротор в современных центрифугах вращается со скоростью до 80000 об/мин, так что на разделяемые частицы действуют силы, превосходящие силу тяготения более чем в 500000 раз. Под действием столь больших сил даже сравнительно небольшие макромолекулы, такие, как тРНК или простейшие ферменты, разделяются и распределяются в строгом соответствии со своими размерами. Измерение коэффициента седиментации макромолекулярных комплексов обычно используют для определения их общей массы и количества входящих в их состав субъединиц.
Ультрацентрифуга разделяет клеточные компоненты не только по массе, но и по плавучей плотности. В этом случае образец седиментирует в крутом градиенте, образованном высококонцентрированным раствором сахарозы или хлористого цезия. Компоненты клеток опускаются по градиенту до тех пор, пока не достигнут участка, плотность раствора в котором равна собственной плотности компонентов. Дальнейшей седиментации компонентов не происходит и они «застревают» на этом уровне. Таким образом в центрифужной пробирке возникает набор различных полос, причем полосы прилежащие к дну пробирки, содержат компоненты максимальной плавучей плотности. Данный метод настолько чувствителен, что с его помощью можно отделять немеченые макромолекулы от макромолекул, содержащих тяжелые изотопы 13С или 15N). Метод центрифугирования в градиенте плотности хлористого цезия был разработан в 1957 году для разделения меченой и немеченой ДНК, синтезированной бактериями в присутствии нуклеотидов, меченных 15N. С помощью этого теперь уже классического эксперимента было показано, что репликация ДНК осуществляется полуконсервативным путем (см. разд. 3.2.3).
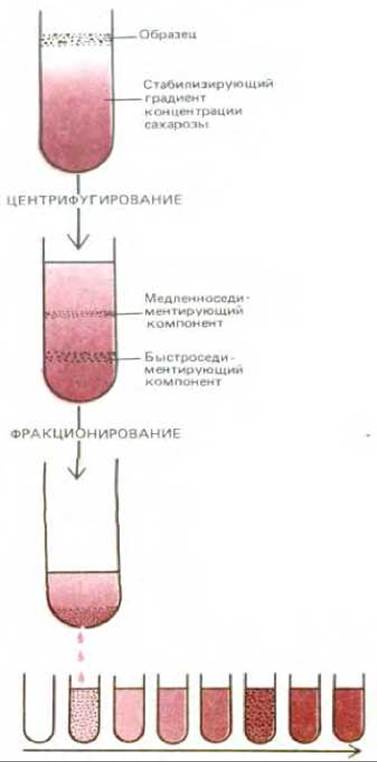
Рис. 4-43. Образец субклеточных компонентов наносят поверх разведенного субклеточного раствора сахарозы и осаждают при различных скоростях в зависимости от размера. В пробирке устанавливается непрерывный градиент плотности сахарозы, концентрация которого возрастает в направлении дна пробирки (обычно используют концентрацию сахарозы в пределах 5-20%). Градиент концентрации сахарозы необходим для стабилизации раствора и седиментирующих полос в условиях конвекции. После центрифугирования различные компоненты можно, как правило, собрать в отдельности. Для этого пластмассовую центрифужную пробирку прокалывают и собирают капли со дна, как это показано на рисунке.
4.4.2. Детали сложных внутриклеточных процессов на молекулярном уровне можно расшифровать в бесклеточных системах [25]
Изучение органелл или других крупных субклеточных компонентов, выделенных с помощью ультрацентрифугирования, чрезвычайно важно для понимания их функций в клетке. Благодаря получению очищенных фракций митохондрий и хлоропластов удалось установить ключевую роль этих органел в процессе превращения энергии. Разделение замкнутых пузырьков, образованных фрагментами гладкого и шероховатого эндоплазматического ретикулума, позволило использовать эти пузырьки в качестве функциональных моделей интактных органелл. Фракционированные клеточные экстракты, называемые также бесклеточными системами, широко используются для изучения внутриклеточных процессов. Только работая с бесклеточными экстрактами можно установить молекулярный механизм биологических процессов, поскольку лишь в этом случае исследуемый механизм может быть изучен в чистом виде - без помех, создаваемых происходящими в клетке побочными реакциями. Использование бесклеточных систем принесло первый триумфальный успех при изучении механизмов биосинтеза белка. Отправной точкой в данном случае послужил неочищенный клеточный экстракт, способный транслировать молекулы РНК в белок. После многократного фракционирования этого экстракта были получены рибосомы, РНК и различные ферменты, составляющие в совокупности аппарат биосинтеза белка. После получения отдельных компонентов в чистом виде их можно было добавлять в систему и исключать из нее и таким образом уточнять роль каждого компонента в процессе биосинтеза белка. Эта же «система трансляции in vitro» оказалась полезной для расшифровки генетического кода - с использованием в качестве матричной РНК (мРНК) искусственных полинуклеотидов известного состава. В настоящее время различные системы трансляции in vitro применяют и для определения механизмов распределения белков по различным внутриклеточным компартментам (см. разд. 8.6.6.), а также для идентификации белков, кодируемых очищенными препаратами мРНК (очистка мРНК является важным этапом в процедуре клонирования генов (см. разд. 5.6.7). В табл. 4-8 приведены некоторые даты из истории разработки методов фракционирования клеточных экстрактов.
Многое из того, что мы знаем о молекулярной биологии клетки открыто при изучении бесклеточных систем. Именно так удалось выяснить механизмы репликации ДНК, транскрипции ДНК, сплайсинга РНК, мышечного сокращения и транспорта частиц по микротрубочкам. Анализ в бесклеточных системах подразумевает полное разделение всех составляющих ее индивидуальных макромолекулярных компонентов и, в частности всех белков, входящих в систему. Методы разделения белков рассматриваются в последующих разделах.
Таблица 4-7. Некоторые типичные коэффициенты седиментации
|
Частица или молекула |
Коэффициент седиментации, S |
|
Лизосома |
9400 |
|
Вирус табачной мозаики |
198 |
|
Рибосома |
80 |
|
Молекула рибосомной РНК |
28 |
|
Молекула тРНК |
4 |
|
Молекула гемоглобина |
4,5 |
Коэффициенты седиментации измеряются в секундах и задаются уравнением (dx/dt)/w2∙ х, где х — расстояние от центра вращения в см, (dx/dt) - скорость осаждения (седиментация) (см/с), ю - угловая скорость вращения ротора центрифуги в радианах в секунду (рад/с). Поскольку такие коэффициенты измеряются очень малыми числами, они обычно выражаются в единицах Сведберга (S), где 1S равен 1 х 10-13 с.
4.4.3. Для фракционирования белков можно использовать хроматографию [26]
В настоящее время хроматография является одним из методов, наиболее широко используемых для фракционирования белков. Первоначально этот метод был разработан для фракционирования низкомолекулярных соединений - Сахаров и аминокислот. Наибольшее распространение получила распределительная хроматография - метод, нашедший широкое применение для разделения небольших молекул. В общей форме этот метод состоит в следующем. Каплю образца наносят на специальную бумагу (хроматография на бумаге) или пластинку стекла или пластмассы, покрытую тонким слоем инертного сорбента, например, целлюлозы или силикагеля (хроматография в тонком слое или тонкослойная хроматография). Затем такую пластинку одним концом помещают в смесь растворителей (например, воды и спирта). По мере движения растворителей по пластинке, они подхватывают те молекулы образца, которые растворяются в них. Растворители выбирают таким образом, чтобы они связывались сорбентом по-разному. В результате молекулы образца, более растворимые в связанном растворителе, движутся медленнее, а другие, более растворимые в слабо сорбированном растворителе, движутся быстрее. Через несколько часов пластинку сушат, окрашивают и определяют положение различных молекул (рис. 4-44).
Белки чаще всего разделяют методом хроматографии на колонках (колоночная хроматография). В этом случае смесь молекул в растворе пропускают через колонку, содержащую твердый пористый матрикс. В результате взаимодействия с матриксом различные белки проходят через колонку с различной скоростью. После того как разные белки достигнут в определенной последовательности дна колонки, их собирают отдельными фракциями (рис. 4-45). В настоящее время разработано и применяется множество матриксов различных типов, используя которые можно делить белки согласно их заряду (ионообменная хроматография), гидрофобности (гидрофобная хроматография), размеру (хроматография гель-фильтрацией) или способности связываться различными химическими группами (аффинная хроматография).
Таблица 4-8. Основные вехи в развитии метода ультрацентрифугирования и приготовления бесклеточных экстрактов
|
1897 - Бюхнер (Buchner) показал, что бесклеточные экстракты дрожжей способны расщеплять сахара с образованием двуокиси углерода и этилового спирта. Так были заложены основы энзимологии 1926 - Сведберг (Svedberg) изобрел аналитическую центрифугу и использовал ее для определения молекулярной массы гемоглобина, которая оказалась равной 68000 дальтон 1935 - Пикелс и Бимс (Pickels, Beams) несколько усовершенствовали конструкцию центрифуги, что позволило использовать ее для проведения препаративных исследований 1938 - Беренс (Behrens) использовал дифференциальное центрифугирование для разделения ядер и цитоплазмы клеток печени. Этот метод был усовершенствован в 40-х и начале 50-х годов Клодом, Браше, Хогебумом (Claude, Brachet, Hageboom) и другими исследователями, что позволило использовать его для разделения органелл клетки 1949 - Сент-Дьердьи (Sent-Geogyi) показал, что изолированные миофибриллы из клеток скелетных мышц сокращаются при добавлении АТР. В 1955 г. аналогичную бесклеточную систему использовал Хофман-Берлинг (Hofmann-Berling) для изучения движения жгутика 1951 - Бракк (Brakke) использовал центрифугирование в градиенте плотности сахарозы для очистки вирусов растений 1954 - де Дюв (de Duve) выделил методом центрифугирования лизосомы, а несколько позже пероксисомы 1954 - Замечник (Samechnik) получил первую бесклеточную систему синтеза белка. За этим открытием последовало десятилетие интенсивных исследований, завершившихся расшифровкой генетического кода 1957 - Мезелсон, Сталь и Виноград (Meselson, Stahl, Vinograd) для разделения нуклеиновых кислот разработали метод центрифугирования в градиенте плотности хлористого цезия |
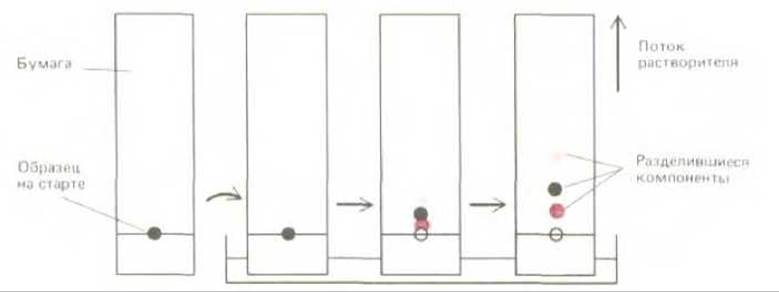
Рис. 4-44. Разделение низкомолекулярных соединений методом хроматографии на бумаге. Образец наносят на старт и высушивают, а затем, используя капиллярный эффект, пропускают сквозь бумагу смесь двух растворителей. Разные компоненты образца движутся по бумаге с различной скоростью, которая зависит от относительной растворимости исследуемых компонентов в растворителе, адсорбируемым бумагой сильнее. Введение этого метода произвело революцию в биохимическом анализе в 40-х годах нашего столетия.
В продаже имеется значительный выбор матриксов различных типов (рис. 4-46). Ионообменные колонки набиты маленькими шариками, заряженными положительно или отрицательно. При использовании таких колонок фракционирование белков происходит в соответствии с расположением зарядов на поверхности белковых молекул. Гидрофобные колонки наполнены шариками, из которых выступают гидрофобные цепи; в таких колонках задерживаются белки с обнаженными гидрофобными участками. Колонки, предназначенные для гель-фильтрации, заполнены крошечными пористыми шариками; при использовании таких колонок происходит разделение белков по размерам. Молекулы небольшого размера по мере прохождения через колонку проникают внутрь шариков, а более крупные молекулы остаются в промежутках между шариками. В результате они быстрее проходят через колонку и выходят из нее первыми. Гель-фильтрация обычно используется и для разделения молекул, и для определения их размеров.
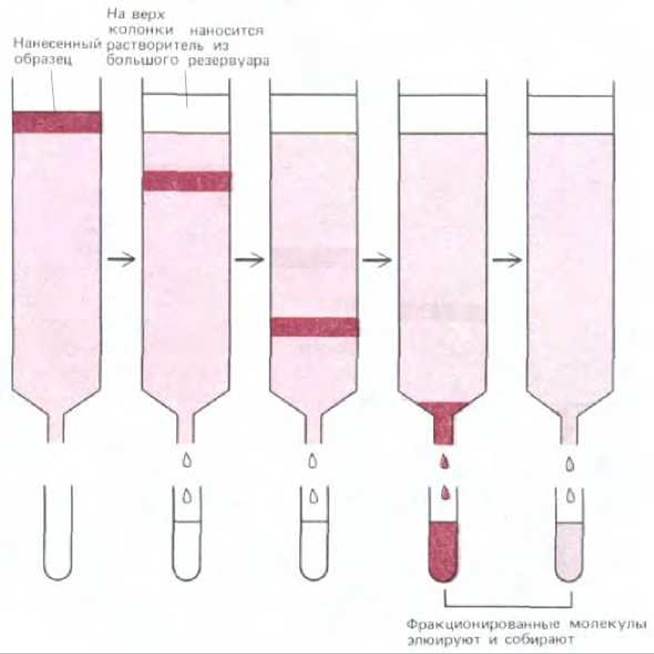
Рис. 4-45. Разделение молекул методом хроматографии на колонках. Образец наносят на верх цилиндрической стеклянной или пластиковой колонки, заполненной проницаемым матриксом (например, целлюлозой), погруженным в растворитель. Затем через колонку медленно прокачивают значительное количество растворителя, который собирают со дна колонки в отдельные пробирки. Различные компоненты образца проходят через колонку с различной скоростью, что и лежит в основе их фракционирования.

Рис. 4-46. Три типа матриксов, используемых для хроматографии. При ионообменной хроматографии (А) нерастворимый матрикс содержит ионы, задерживающие молекулы с противоположным зарядом. Для разделения молекул используются следующие матриксы: диэтиламиноэтилцеллюлоза (ДЭАЭ-целлюлоза) - заряжена положительно; карбоксиметилцеллюлоза (КМ-целлюлоза) и фосфоцеллюлоза - заряжены отрицательно. Силы взаимодействия между молекулами в растворе и ионообменником определяются ионной силой и рН элюирующего раствора, которые для достижения эффективного разделения можно варьировать определенным образом (как на рис. 4-47). При хроматографии по методу гель- фильтрации (Б) матрикс инертен, но содержит поры. Низкомолекулярные соединения проникают внутрь частиц матрикса. Оказавшись при этом в относительно большем объеме, они проходят через колонку медленнее. В качестве матрикса можно использовать зерна поперечно-сшитого полисахарида (декстран или агароза). Поскольку в продаже имеются полисахариды с самым различным размером пор, их можно использовать для фракционирования молекул с молекулярной массой от 500 до 5 х 106 дальтон. При аффинной хроматографии (В) используется нерастворимый матрикс, ковалентно связанный со специфичными лигандами (антителами или субстратом ферментов), которые присоединяют определенный белок. Связываемые иммобилизованным субстратом молекулы фермента можно элюировать концентрированными растворами субстрата в свободной форме, а молекулы, связанные с иммобилизованными антителами, можно элюировать за счет диссоциации комплекса антитело антиген концентрированными растворами соли или растворами низкого или высокого рН. Однократная хроматография на такой колонке позволяет зачастую достигнуть очень высокой степени очистки препарата.
На каждом этапе колоночной хроматографии содержание белка в смеси увеличивается не более, чем в 20 раз, и поэтому выделить из сложной смеси белков отдельный белок за один цикл практически невозможно. На долю каждого белка, как правило, приходится менее 1/1000 всего белка клетки, и для его очистки требуется последовательное использование нескольких различных типов колонок (рис. 4-47). Гораздо более эффективен метод аффинной хроматографии (хроматография по сродству). В основе этого метода лежат биологически важные взаимодействия, происходящие на поверхности белковых молекул. Так, при ковалентном связывании субстрата фермента с матриксом, например, с полисахаридными шариками, фермент специфически удерживается матриксом и может быть элюирован (смыт) практически в чистом виде. Подобным образом можно иммобилизировать короткие олигонуклеотиды ДНК определенной структуры (см. разд. 4.6.8) и использовать подобные носители для очистки ДНК-связывающих белков, опознающих данную последовательность нуклеотидов на хромосомах (см. разд. 9.1.8). С матриксом можно связать и специфические антитела; такой носитель очень удобен для очистки белков, узнаваемых этими антителами. Аффинные колонки обладают высокой степенью специфичности; за один цикл хроматографии можно добиться очень высокой степени очистки (1000-10000 раз).
Разрешение обычной колоночной хроматографии ограничено негомогенностью матриксов (например, целлюлозы), что вызывает неравномерное протекание растворителя через колонку. Разработанные недавно хроматографические смолы (в основу которых обычно положен кремний) имеют форму мельчайших сфер от 3 до 10 мкм в диаметре, которые упакованы в специальный чехол и образуют гомогенную колонку. Такие колонки для высокоэффективной жидкостной хроматографии (ВЖХ) обеспечивают высокий уровень разрешения.
Поскольку частицы носителя в колонках для ВЖХ упакованы очень плотно, в отсутствие высокого давления скорость потока через них незначительна. По этой причине такие колонки обычно помещают в стальные цилиндры, соединенные со сложной системой насосов и шлангов, которые обеспечивают необходимое для высокой скорости протока давление. В традиционной колоночной хроматографии скорость протекания через колонку может быть довольно низкой (примерно один объем колонки в час), таким образом, у разделяемых растворов достаточно времени для уравновешивания с внутренним содержимым крупных частиц матрикса. В условиях ВЖХ происходит быстрое уравновешивание растворов с внутренним содержимым крошечных сфер, так что растворы, обладающие различным сродством к матриксу, эффективно разделяются даже при высокой скорости потока. Таким образом, ранее для достижения плохого разделения с помощью колоночной хроматографии требовались часы, а в настоящее время благодаря ВЖХ качественное фракционирование занимает минуты. Вот почему именно этот метод чрезвычайно популярен сейчас для разделения и белков, и малых молекул.
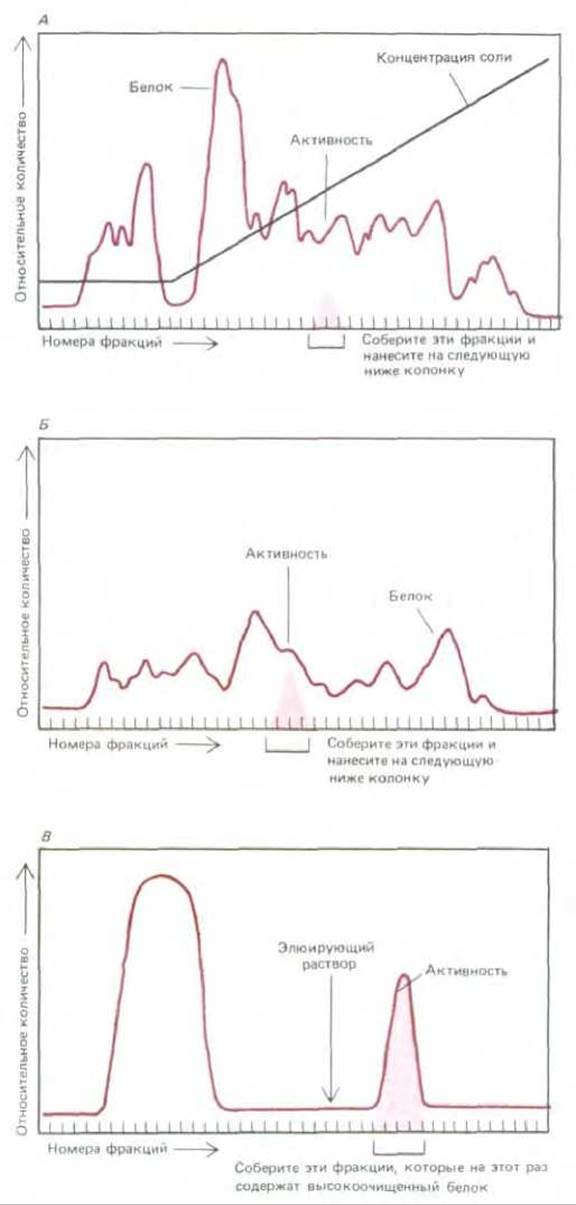
Рис. 4-47. Типичные результаты, полученные при очистке белка различными методами хроматографии. В данном случае подлежащий фракционированию клеточный экстракт сначала пропускали через колонку, заполненную ионообменной смолой (А). Затем колонку промывали и связавшиеся белки элюировали раствором, содержащим постепенно нарастающую концентрацию соли. Белки с наименьшим сродством к ионообменной смоле проходят через колонку не задерживаясь и собираются со дна колонки в первых порциях элюата. Остальные белки элюируются соответственно сродству к ионообменной смоле. Для элюирования белков, связывающихся со смолой наиболее сильно, требуется наивысшая концентрация соли. Исследуемый белок элюировался в виде узкого пика; он был выявлен по ферментативной активности. Фракции с такой активностью собирали и наносили на вторую колонку для гель-фильтрации (Б). Фракцию все еще недостаточно очищенного белка выявляли по ферментативной активности; активные фракции собирали и очищали до гомогенного состояния на колонке (В), содержащей иммобилизованный субстрат фермента.
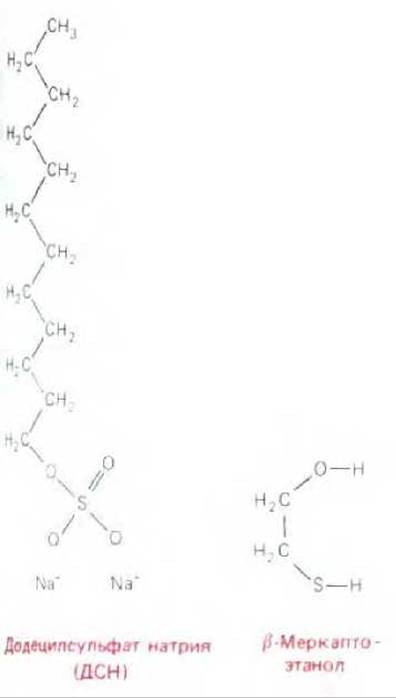
Рис. 4-48. Детергент додецилсульфат натрия (ДСН) в ионизированной форме и восстановитель ß-меркаптоэтанол. Эти два реактива используются для солюбилизации белков при ДСН-электрофорезе в полиакриламидном геле.
4.4.4. С помощью электрофореза в полиакриламидном геле в присутствии додецилсульфата натрия (ДСН) можно определить размеры и субъединичный состав белков [27]
Белки обычно несут суммарный положительный или отрицательный заряд, обусловленный наличием на их поверхности положительно или отрицательно заряженных групп аминокислот. Если белковые молекулы поместить в электрическое поле, они начинают перемещаться со скоростью, которая определяется их суммарным зарядом, а также формой и размерами. Этот феномен лежит в основе электрофореза - метода разделения смесей белков в свободных водных растворах и в твердом пористом матриксе, в качестве которого можно использовать крахмал.
В середине 60-х годов был разработан модифицированный метод электрофореза - электрофорез в полиакриламидном геле в присутствии додецилсульфата натрия (ДСН-ПААГ). Этот метод был существенным шагом вперед по сравнению с обычными методами анализа белков, известными к тому времени. При использовании данного метода белки мигрируют в инертном матриксе - полиакриламидном геле с высоким содержанием поперечных сшивок. Обычно гель готовят полимеризацией мономеров непосредственно перед использованием. Размеры пор геля могут быть подобраны произвольно с тем, чтобы гель мог замедлить миграцию определенных молекул. При этом белки находятся в растворе, содержащем мощный, отрицательно заряженный детергент - додецил-сульфат натрия или ДСН (SDS) (рис. 4-48).
Связываясь с гидрофобными участками белковой молекулы, этот детергент вызывает развертывание белковых молекул в длинные вытянутые цепи. Развертываясь, отдельные белковые молекулы освобождаются из комплексов с белками или молекулами липидов и солюбилизируются в растворе детергента. В качестве восстанавливающего агента обычно добавляют меркаптоэтанол (рис. 4-48), разрушающий в белках связи S-S. Это дает возможность анализировать полипептиды, образующие мультисубъединичные молекулы.
Что же произойдет, если смесь белков, растворенных в ДСН, подвергнуть электрофорезу в блоке полиакриламидного геля. Каждая молекула белка связывает значительное количество негативно заряженных молекул детергента, общий заряд которых превосходит общий заряд белка. По этой причине белок после того, как будет приложено напряжение, начнет двигаться в направлении положительного электрода. Белки одного размера ведут себя сходным образом, поскольку, во-первых, их природная структура полностью нарушена ДСН так, что их форма идентична, во-вторых, они связывают одинаковое количество ДСН и приобретают одинаковый негативный заряд. Крупные белки, обладающие большим зарядом, подвергаются действию значительных электрических сил, а также более существенному торможению. В обычных растворах эти эффекты, как правило, взаимно погашаются, но в порах полиакриламидного геля, действующего как молекулярное сито, большие белки тормозятся значительно сильнее, чем малые белки. Вследствие этого сложная смесь белков делится на ряд полос, расположенных в соответствии с их молекулярной массой. Окрасив гель красителем кумасси синим, можно выявить основные фракции полипептидов. Минорные белки идентифицируют серебрением; минимальное количество белка, выявляемое в полосе, составляет в последнем случае 10 нг. С помощью таких гелей можно идентифицировать специфический белок, если пометить его антителами, связанными с радиоактивными изотопами, ферментами или флуоресцирующими красителями. Идентификацию часто выполняют после переноса белков из геля на лист нитроцеллюлозы (посредством «блоттинга»). Ниже этот метод описан более подробно применительно к изучению нуклеиновых кислот (см. разд. 4.6.8). Описанный метод выявления белка назван вестерн-блоттингом.
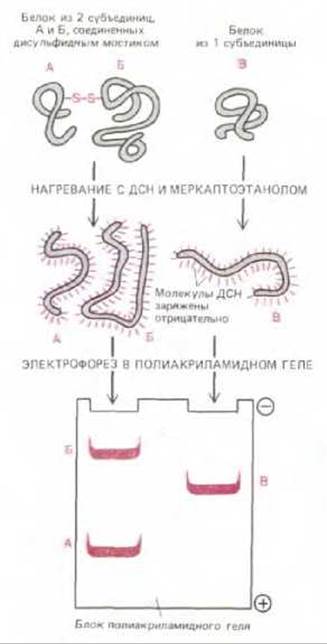
Рис. 4-49. ДСН-электрофорез в полиакриламидном геле. Индивидуальные белки образуют комплекс с молекулами додецилсульфата натрия, несущими отрицательный заряд, и мигрируют через пористый гель полиакриламида в виде отрицательно заряженного комплекса ДСН-белок. Поскольку скорость передвижения в этих условиях тем выше, чем меньше размеры полипептида, этот метод может быть использован для определения приблизительной молекулярной массы полипептидной цепи, а также для изучения субъединичного состава белка.
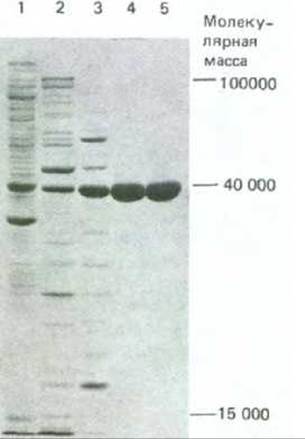
Рис. 4-50. Анализ образцов белка методом электрофореза в ДСН-полиакриламидном геле. На фотографии показан гель, использованный для выявления белков, присутствующих на последующих стадиях очистки фермента. Самая левая дорожка (дорожка 1) содержит сложную смесь белков исходного клеточного экстракта, каждая из последующих дорожек содержит белки, полученные после хроматографического фракционирования белковых образцов, анализированных на предыдущей дорожке (см. рис. 4-47). В лунку каждой дорожки на гель наносили одинаковое количество белка (10 мкг). Отдельные белки в норме проявляются в виде узких окрашенных полос; полосы расширяются, если в них присутствует слишком много белка. (С любезного разрешения Tim Formosa.)
Метод ДСН-электрофореза белков в полиакриламидном геле значительно мощнее любого другого метода фракционирования белков из известных ранее хотя бы потому, что может быть использован для выявления любого белка независимо от его растворимости в воде. С помощью этого метода можно разделить на отдельные фракции белки мембран, белковые компоненты цитоскелета и белки, входящие в состав крупных макромолекулярных агрегатов. При использовании этого метода полипептиды разделяются строго по размеру, поэтому с его помощью можно получить информацию о субъединичном составе любого комплекса и о молекулярной массе белков, образующих этот комплекс (рис. 4-49). Фотография геля, который был использован для анализа последовательных этапов очистки белка, представлена на рис. 4-50.
4.4.5. Методом двумерного гель-электрофореза можно разделить в одном геле более 1000 белков [28]
Известно, что близко расположенные полосы в геле могут перекрываться. Этот эффект препятствует выявлению большого количества белков (не больше 50) с помощью одномерных методов их разделения, Метод двумерного гель-электрофореза, в котором объединены две различные процедуры разделения, позволяет идентифицировать более 1000 белков. Результаты при этом получают в виде «двумерной» белковой карты.
При работе данным методом на первом этапе белки разделяют по их заряду. Для этого образец помещают в небольшой объем раствора, содержащего неионный (незаряженный) детергент - меркаптоэтанол, и в качестве денатурирующего агента - мочевину. В этом растворе происходит солюбилизация, денатурация и диссоциация всех без исключения полипептидных цепей; при этом изменения заряда цепей не происходит. Диссоциированные полипептидные цепи разделяют затем методом изоэлектрического фокусирования, основанном на изменении заряда белковой молекулы при изменении рН окружающей среды. Каждый из белков может быть охарактеризован изоэлектрической точкой - значением рН, при котором суммарный заряд белковой молекулы равен нулю, и, следовательно, белок не способен перемещаться под действием электрического поля. При изоэлектрическом фокусировании белки подвергаются электрофорезу в узкой трубочке, заполненной полиакриламидным гелем, в котором с помощью специальных буферов создается градиент рН. Под действием электрического поля каждый белок перемещается в ту зону градиента, которая соответствует его изоэлектрической точке и остается в ней (рис. 4-51). Так происходит разделение белков в одном направлении двумерного гель-электрофореза.
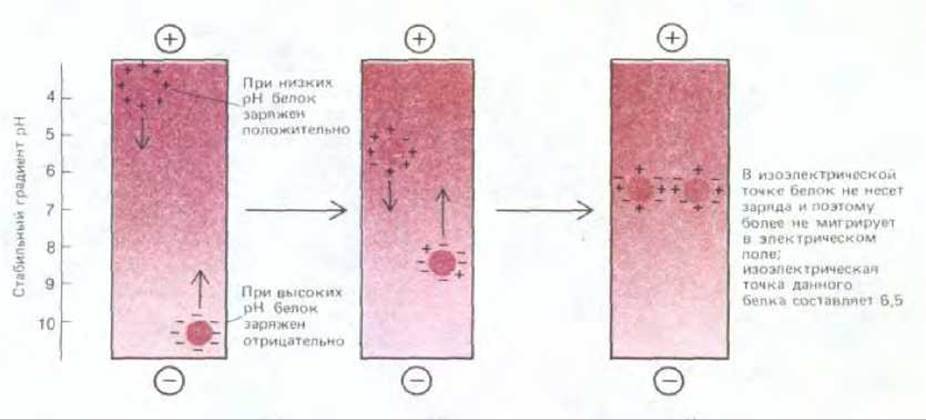
Рис. 4-51. Разделение молекул белка методом изоэлектрического фокусирования. При низких значениях рН (высокое содержание ионов Н+) карбоксильные группы белков имеют тенденцию оставаться незаряженными (—СООН), а основные, азотсодержащие группы белков полностью заряжены (например, -NH3+), что обусловливает у белков суммарный положительный заряд. При высоких значениях рН карбоксильные группы заряжены отрицательно (—COO-), а основные группы имеют тенденцию оставаться незаряженными, например (NH2). В результате белки приобретают отрицательный суммарный заряд (см. рис. 2-8). При изоэлектрической точке белок незаряжен, поскольку положительный и отрицательный заряды уравновешены. Следовательно, если пробирку, содержащую раствор с фиксированным градиентом рН, подвергнуть действию сильного электрического поля, каждый вид белка будет перемещаться до тех пор, пока не образует узкой полосы в зоне рН, соответствующего изоэлектрической точке, как показано на рисунке.
На втором этапе трубочка геля, содержащего разделенные белки, снова подвергается электрофорезу, на этот раз в направлении перпендикулярном тому, что на первом этапе. В этом случае электрофорез ведут в присутствии ДСН и белки разделяют по их молекулярной массе, как в одномерном ДСН-ПААГ. Исходный гель пропитывают додецил-сульфатом натрия и, поместив его на блок ДСН-ПААГ-геля, проводят электрофорез, в ходе которого каждая из полипептидных цепей мигрирует сквозь блок геля и образует в нем отдельную полосу. Так осуществляется разделение во втором направлении двумерного гель-электрофореза. Неразделенными в результате остаются только те белки, которые неразличимы как по изоэлектрической точке, так и по молекулярной массе; такое сочетание встречается очень редко.
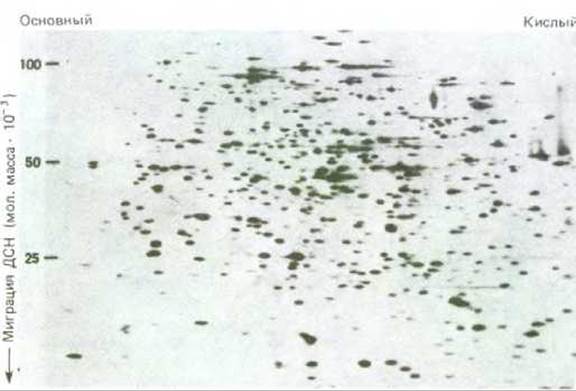
Рис. 4-52. Фракционирование белков клетки Е. coli методом двумерного электрофореза в полиакриламидном геле. Каждое пятно соответствует отдельной полипептидной цепи. Сначала белки разделяли соответственно их изоэлектрическим точкам методом изоэлектрического фокусирования слева направо. Затем в присутствии ДСН их разделяли методом электрофореза сверху вниз в соответствии с молекулярной массой их субъединиц. Отметим, что содержание разных белков в клетке неодинаково. (С любезного разрешения Patrick O'Farrell.)
Таблица 4-9. Основные вехи в развитии методов хроматографии и электрофореза и в применении этих методов для разделения биологических макромолекул
|
1833 - Фарадей (Faradey) сформулировал фундаментальные законы, описывающие электрические явления в растворах 1850 - Рунге (Rounge) разделил неорганические соединения по их дифференциальной адсорбции на бумаге, предвосхитив тем самым появление методов хроматографического разделения 1906 - Цвет изобрел хроматографию на колонках. Он пропустил петролейные экстракты листьев растений через колонку с порошкообразным мелом 1933 - Тизелиус (Thiselius) использовал электрофорез для разделения белков в растворе 1942 - Мартин и Синж (Martin, Synge) изобрели распределительную хроматографию, на основе которой через два года был разработан метод хроматографии на бумаге 1946 - Стайн и Мур (Stain, Moore) впервые определили аминокислотный состав белка. Первыми в качестве наполнителя в колоночной хроматографии они использовали крахмал, а позже ионообменные смолы 1955 - Смитис (Smithies) для разделения белков с помощью электрофореза использовал крахмальный гель 1955 - Сэнгер (Sanger) завершил анализ аминокислотной последовательности бычьего инсулина. Это первый белок, у которого определена полная аминокислотная последовательность 1956 - Ингрэм (Ingram) получил первые пептидные карты («фингерпринты» - «отпечатки пальцев»), показав при этом, что различия гемоглобина больных серповидноклеточной анемией и нормального гемоглобина обусловлены заменой одной-единственной аминокислоты 1959 - Рэймонд (Raymond) ввел в лабораторную практику полиакриламидный гель, который превосходит гель из крахмала при электрофоретическом разделении белков; в течение нескольких последующих лет Орнстайн и Дэвис (Ornstain, Davis) разработали более эффективные буферные системы, что позволило проводить разделение белков с высокой степенью разрешения 1966 - Мэйзел (Mayzel) для усовершенствования разделения белков в полиакриламидном геле предложил использовать додецилсульфат натрия (ДСН-SDS) 1975 - О’Фаррел (O'Farrell) разработал систему двумерного гель-электрофореза для анализа белковых смесей. Предложенный им метод представляет собой сочетание ДСН-электрофореза белков в полиакриламидном геле и изоэлектрического фокусирования 1984 - Шварц и Кантор (Schwartz, Cantor) разработали метод электрофореза в пульсирующем электрическом поле (пульс-электрофорез), используемый для разделения очень больших молекул ДНК |
Используя различные методы окрашивания белков, а в случае радиоактивно меченных белков - метод радиоавтографии (см. разд. 4.5.2), можно выявить следовые количества практически всех полипептидных цепей. За один раз методом двумерного гель-электрофореза можно разделить до 2000 отдельных полипептидных цепей; этого достаточно, чтобы выявить большинство бактериальных белков (рис. 4-52). Разрешение этого метода настолько велико, что позволяет разделить два практически идентичных белка, отличающихся одной заряженной аминокислотой. Таблица 4-9 знакомит нас с основными этапами развития методов хроматографии и электрофореза.
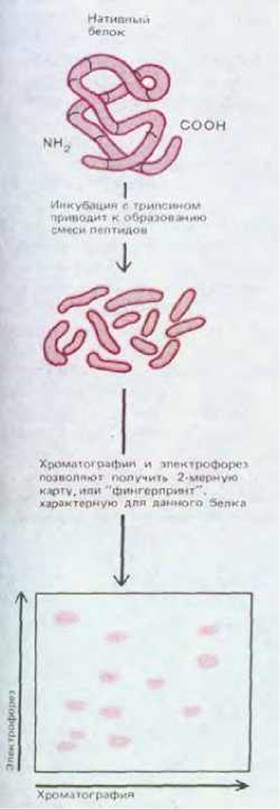
Рис. 4-53. Получение пептидной карты («фингерпринта» или «отпечатков пальцев») белков. В данном случае белок расщепляли трипсином и получили смесь мелких фрагментов полипептидов. Эту смесь фракционировали в двух направлениях: электрофорезом и распределительной хроматографией. Полученная картина пятен характеризует данный белок.
4.4.6. Избирательное расщепление белка приводит к образованию характерного набора пептидных фрагментов [29]
Молекулярная масса и изоэлектрическая точка - характерные параметры белка. Однако в основе точной идентификации белковой молекулы лежит определение аминокислотной последовательности. Уже на первом этапе этого процесса, включающего расщепление белка на мелкие фрагменты, можно получить значительную информацию о данном белке. В настоящее время в продаже имеются протеолитические ферменты и химические реактивы, расщепляющие белки по определенным аминокислотным остаткам (табл. 4-10). Так, фермент трипсин отщепляет остатки лизина и аргинина со стороны карбоксильных групп; химический реактив бромистый циан расщепляет пептидные связи, расположенные после остатков метионина. Поскольку такие специфические ферменты и реактивы расщепляют в белковой молекуле ограниченное количество связей, при их воздействии образуется смесь больших пептидов. Разделив эту смесь методом электрофореза или хроматографии, можно получить пептидную карту, характеризующую исследуемый белок. Такие пептидные карты называют иногда «фингерпринтами» (отпечатками пальцев) белка (рис. 4-53).
Таблица 4-10. Некоторые реактивы, используемые для расщепления пептидных связей в белках
|
Аминокислота 1 |
Аминокислота 2 |
|
|
Фермент |
||
|
Трипсин |
Лизин или аргинин |
Любая |
|
Химотрипсин |
Фенилаланин, триптофан или тирозин |
» |
|
V8-Протеаза |
Глутаминовая кислота |
» |
|
Химический реактив |
||
|
Бромистый циан |
Метионин |
» |
|
2-Нитро-5-тиоцианобензоат |
Любая |
Цистеин |
Указана специфичность в отношении аминокислот с каждой стороны от расщепляемой связи. После расщепления высвобождается карбоксильная группа по аминокислоте 1; эта аминокислота расположена слева от пептидной связи при нормальном написании (см. схему 2-5).
Этот метод был разработан в 1956 г. для сравнения нормального гемоглобина с мутантной формой того же белка, обнаруживаемой в крови больных серповидноклеточной анемией. Оказалось, что мутантный белок отличается от нормального по одной-единственной аминокислоте. Так впервые было доказано, что мутация может привести к замене в белке только одной аминокислоты.
4.4.7. С помощью автоматических приборов можно анализировать короткие аминокислотные последовательности [30]
Осуществив расщепление белка на мелкие фрагменты, приступают к следующему этапу - определяют последовательность аминокислот в каждом из выделенных пептидных фрагментов. Для этого проводят серию химических реакций, которые впервые были предложены в 1967 году. Сперва пептид обрабатывают каким-либо реактивом, взаимодействующим только со свободной аминогруппой на его N-конце. Далее этот реактив активируют, воздействуя на него слабой кислотой. Теперь он специфически расщепляет пептидную связь, соединяющую N-концевую аминокислоту с пептидной цепью; высвобождающуяся при этом аминокислоту идентифицируют методом хроматографии. Оставшийся пептид укорачивается в результате на одну аминокислоту. Его также подвергают реакциям, проводимым в той же последовательности, - и так, пока в пептиде не будет определена каждая аминокислота.
Циклический характер этих реакций дает возможность автоматизировать весь процесс. В настоящее время выпускаются приборы (аминокислотные секвенаторы), производящие автоматическое определение последовательности аминокислот в пептидных фрагментах. На последнем этапе анализа последовательности аминокислот, полученные для пептидных фрагментов, располагают в том же порядке, как они были расположены в интактной цепи. Для этого сравнивают последовательности наборов перекрывающихся фрагментов, полученных при расщеплении одного и того же белка различными протеолитическими ферментами.
Усовершенствование техники секвенирования белка значительно повысило его скорость и чувствительность, позволяя анализировать минимальные количества образца. Например, в настоящее время последовательность из нескольких десятков аминокислот можно выяснить, имея в распоряжении всего несколько микрограммов белка - количество, извлекаемое из одной полосы ДСН-полиакриламидного геля. Это оказалось крайне важно для изучения многих минорных белков клетки, например, рецепторов стероидных или полипептидных гормонов. В настоящее время достаточно определить в белке 20 аминокислот, чтобы сконструировать ДНК-зонд, используемый для клонирования соответствующего гена (см. разд. 5.6.5). После выделения гена оставшаяся невыясненной часть аминокислотной последовательности белка может быть реконструирована по нуклеотидной последовательности согласно генетическому коду. Это можно считать значительным достижением, поскольку даже с полной автоматизацией определение полной первичной последовательности белка остается крайне сложной задачей. Так, например, если белок состоит из 100 аминокислот, их последовательность, если очень напряженно трудиться, можно установить за месяц. Но с удлинением цепи аминокислот сложности нарастают очень быстро, что не позволяет превратить процесс определения аминокислотной последовательности в рутинную методику. Учитывая то обстоятельство, что секвенирование ДНК - процедура более легкая и занимает меньше времени (см. ниже), в настоящее время последовательность аминокислот в большинстве белков, как правило, определяют по нуклеотидной последовательности соответствующих генов.
Заключение
Клеточные популяции можно анализировать биохимически, разрушая клетки и анализируя их содержимое с помощью ультрацентрифугирования. Дальнейшее фракционирование позволяет создать функциональные бесклеточные системы; такие системы необходимы для определения молекулярных деталей сложных клеточных процессов. Например, с помощью этого метода в недавнее время были исследованы синтез белка, репликация ДНК, сплайсинг РНК и различные типы внутриклеточного транспорта.
Мажорные белки растворимых клеточных экстрактов можно очищать с помощью колоночной хроматографии; в зависимости от типа матрикса в колонках биологически активные белки можно разделять по их молекулярной массе, гидрофобности, характерному заряду либо сродству с иными молекулами. В ходе очистки, как правило, образец пропускают через несколько колонок - обогащенные фракции, полученные с одной колонки, наносят на следующую. После очистки белка до гомогенного состояния проводят тщательное определение его биологической активности. Можно определить также небольшой фрагмент аминокислотной последовательности белка и клонировать его ген; оставшуюся часть аминокислотной последовательности реконструируют по последовательности нуклеотидов в гене.
Таблица 4-11. Использование некоторых радиоактивных изотопов в биологических исследованиях
|
Изотоп |
Период полураспада |
|
32р |
14 сут |
|
131I |
8,1 сут |
|
35S |
87 сут |
|
14С |
5570 лет |
|
45Са |
164 сут |
|
3Н |
12,3 года |
1) Изотопы расположены в порядке уменьшения энергии испускаемых ими электронов. 131I испускает также у-лучи. Период полураспада - время, в течение которого распадается 50% атомов данного изотопа.
Даже если количество белка очень невелико, его молекулярную массу и субъединичный состав можно определить, используя ДСН- электрофорез в ПААГ. В случае двумерного электрофореза белки разделяют на отдельные фракции изоэлектрическим фокусированием в одном направлении, после чего следует ДСН-электрофорез во втором направлении. Этот метод может быть использован для разделения тех белков, которые в норме считаются нерастворимыми.