Молекулярная биология клетки - Том 3 - Албертс Б., Брей Д., Льюис Дж., Рэфф М., Робертс К., Уотсон Дж. 1994
От клеток к многоклеточным организмам
Рак
Молекулярная генетика рака
Поскольку рак - результат серии случайных генетических событий, вряд ли найдутся хотя бы две опухоли даже одного вида, которые были бы генетически идентичны. Несмотря на это, можно ожидать, что при любой форме рака нарушаются нормальные ограничения пролиферации клеток, и для каждого типа клеток существует определенное число возможных способов реализации подобного нарушения. Более того, некоторые элементы механизма, регулирующего клеточное деление, по-видимому, одинаковы во многих или даже во всех типах клеток, и одинаково уязвимы. Фактически основной вклад в нарушение регуляции деления клеток при раке вносит относительно небольшое число генов. Идентификация и характеристика многих из них - одно из крупнейших достижений молекулярной биологии за последнее десятилетие. Пролиферация клеток может регулироваться непосредственно - через механизм, заставляющий клетку начинать очередной цикл деления (см. разд. 13.3.2), или косвенно - например, через регуляцию вступления клетки на путь терминальной дифференцировки (см. разд 17.4.1). В обоих случаях нормальные регуляторные гены можно разделить на две категории - те, продукты которых способствуют стимуляции пролиферации клеток, и те, чьи продукты участвуют в ее торможении. Соответственно, есть два вида мутаций, ведущих к неконтролируемой пролиферации - основному свойству раковых клеток. Мутации первого типа приводят к гиперактивности «стимулирующего» гена; они доминантны (для проявления достаточно мутации в одной из двух клеточных копий такого гена), а измененный ген называется онкогеном (его нормальный аллель - протоонкогеном). Мутации второго типа приводят к инактивации «ингибирующего» гена; эти мутации рецессивны - обе клеточные копии гена должны быть инактивированы или удалены, чтобы освободить клетку от ингибирующего контроля; утерянный ген называют иногда геномсупрессором опухолевого роста, или опухолевым супрессорным геном. Помимо обычных мутаций существуют генетические изменения другого типа, которые также могут приводить к развитию рака - система контроля клеточного деления может быть разрушена чужеродной ДНК, которая вводится в клетку вирусом. В действительности изучение молекулярной генетики рака началось именно с открытия таких опухолеродных вирусов, что подготовило почву для последующего обнаружения онкогенов и протоонкогенов. Успехи в решении другой (и более трудной) задачи - идентификации и клонировании опухолевых супрессорных генов - были достигнуты сравнительно недавно (о них пойдет речь в конце этого раздела).
21.2.1. Опухоли могут вызываться как ДНК-, так и РНК-содержащими вирусами [15, 16]
И ДНК-, и РНК-содержащие вирусы (в частности, ретровирусы) могут участвовать в трансформации нормальной клетки в опухолевую. Это можно экспериментально продемонстрировать как на лабораторных животных (у которых некоторые вирусы способны вызывать рак), так и в культуре клеток, где те же вирусы изменяют поведение инфицированных клеток. Эти клетки приобретают способность к делению в условиях, при которых нормальные клетки делиться не могут (см. разд. 13.4.1, где перечислены свойства неопластически трансформированных клеток в культуре). Два интенсивно изучаемых примера таких вирусов - это SV40 (ДНК-содержащий вирус, выделенный из клеток обезьяны) и вирус саркомы Рауса - куриный ретровирус. Сложнее обстоит дело с ролью вирусов в развитии рака у человека; среди множества причин, приводящих практически ко всем известным видам раковых заболеваний человека, вирусы не фигурируют. Возможно, от многих вирус-индуцированных опухолей нас защищает иммунная система, разрушающая инфицированные вирусами клетки, которые могли бы стать источником опухолей. Тем не менее сейчас имеются веские доказательства того, что причиной возникновения некоторых типов рака человека являются вирусы (табл. 21-3). Они могут оказывать либо непрямое промоторное действие, либо способствовать неопластической трансформации инфицированных клеток.
В устойчиво трансформированной вирусом клетке должно установиться равновесие, вирус не должен убивать клетку, а клетка должна сохранять вирусные гены при размножении (передавать из поколения в поколение). Обычно это осуществляется путем интеграции этих генов в одну или более клеточных хромосом. Но иногда вирусные гены существуют в клетке в виде плазмиды, реплицирующейся одновременно с хромосомами. Так, вирус SV40 и ретровирусы встраиваются в хромосомы клеток, которые они трансформируют; папилломавирусы - класс ДНК-содержащих вирусов, вызывающих у человека образование бородавок (и, вероятно, участвующих в возникновении рака шейки матки), при одних условиях существуют в виде плазмид, при других - как интегрированные элементы. В обоих случаях они «наделяют» клетку наследственно измененным геномом. Однако ДНК-содержащие вирусы и ретровирусы отличаются друг от друга по природе генов, вызывающих неопластическую трансформацию, и по тому, какое место в обычном жизненном цикле вируса занимает трансформация клетки.
Таблица 21-3. Вирусы, связанные с раковыми заболеваниями у человека
|
Вирус |
Связанные с ним опухоли |
Районы с высокой заболеваемостью |
Другие предполагаемые факторы риска |
|
ДНК-содержищие вирусы |
|||
|
Семейство паповавирусов |
|||
|
Папилломавирус (разнообразные штаммы) |
Бородавки (доброкачественные) |
Повсюду |
- |
|
рак шейки матки |
Повсюду |
Курение |
|
|
Семейство гепаднавирусов |
|||
|
Вирус гепатита В |
Рак печени (гепатоклеточная карцинома) |
Юго-Восточная Азия, тропическая Африка |
Афлатоксин (при заражении пищи грибками), алкоголизм, курение, другие вирусы |
|
Семейство герпесвирусов Вирус Эпштейна-Барр |
Лимфома Беркитта (рак В-лимфоцитов) |
Западная Африка, Папуа - Новая Гвинея Южный |
Малярия |
|
Носоглоточный рак |
Китай, Гренландия (инуиты) |
Генотип гистосовместимости (?), питание соленой рыбой в детстве (?) |
|
|
РНК-содержащие вирусы |
|||
|
Семейство ретровирусов |
|||
|
Вирус Т-лейкоза человека типа I (HTLV-І) |
Т-клеточный лейкоз/лимфома у взрослых |
Япония (Кюсю), Вест-Индия |
- |
|
Вирус иммунодефицита человека (HIV-1, вирус СПИДа) |
Саркома Капоши (рак эпителиальных клеток кровеносных сосудов?) |
Центральная Африка |
Иммунная недостаточность или супрессия, заражение другим вирусом (?) |
Для всех указанных вирусов число инфицированных людей гораздо больше числа заболевших раком: вирусы должны действовать в сочетании с другими факторами. Более того, вклад некоторых вирусов в возникновение рака только косвенный: вирус СПИДа, например, подавляет клеточную иммунную защиту; в результате клетки эндотелия, которые были трансформированы какими-то другими агентами, не уничтожаются иммунной системой, а образуют опухоль.____________________________________
21.2.2. Нарушение контроля клеточного деления ДНК-содержащими онкогенными вирусами - часть их стратегии выживания [15, 17]
Как уже сообщалось в гл. 5 (разд. 5.5.3), размножение опухолеродных ДНК-содержащих вирусов, таких как SV40, в естественных условиях не сопровождается развитием рака. Проникнув в клетку хозяина, SV40 обычно жестко не встраивается в клеточный геном. Вместо этого кодируемый вирусным геном белок (или группа белков) быстро активирует клеточную систему репликации ДНК, и затем вирус использует ее для репликации собственной ДНК, которая в свою очередь служит матрицей для синтеза других компонентов вируса за счет клетки хозяина. Этот процесс производства вирусных частиц клеткой продолжается до тех пор, пока она не погибнет, высвобождая множество новых вирусов. Значительно реже вирус попадает в непригодную для его размножения клетку, где может пребывать сколь угодно долгое время в результате устойчивого внедрения в одну или более клеточных хромосом. В этом случае вирусный ген, ответственный за активацию репликации клеточной ДНК, также может транскрибироваться, подталкивая таким образом, клетку к вступлению в S-фазу клеточного цикла и заставляя ее снова и снова совершать цикл деления. Вирусный ген начинает работать как онкоген, вызывая опухолевую трансформацию. Однако такой онкоген принципиально отличается от тех классов онкогенов, которые мы рассмотрим ниже, - у него нет гомолога в геноме нормальной клетки.
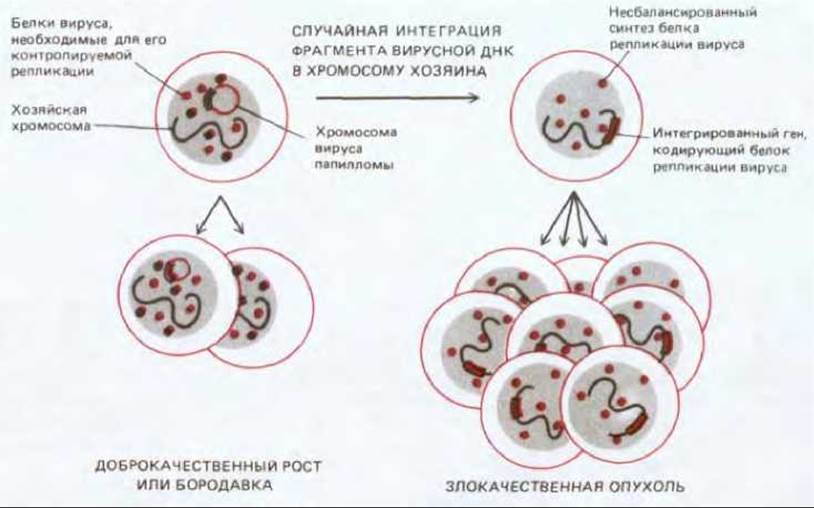
Рис. 21-20. Предполагаемый механизм, с помощью которого определенные папилломавирусы могут индуцировать рак шейки матки. Данные вирусы имеют кольцевую двухцепочечную ДНК длиной около 8000 пар оснований. В клетках бородавок и других доброкачественных образований вирусные хромосомы стабильно существуют в виде плазмид и реплицируются самостоятельно. В результате редкого случайного события вирусный геном может встроиться в хромосому хозяина. При этом изменится окружение вирусных генов и нарушится контроль их экспрессии. Нерегулируемый синтез белка репликации вируса «подталкивает» клетку к вступлению в S-фазу клеточного цикла, способствуя тем самым возникновению рака.
ДНК-содержащие вирусы - весьма разнообразная группа, но описанные общие принципы, с некоторыми изменениями (вариациями), применимы к большинству из них, вовлеченных в патогенез рака. Примером одного из вариантов являются папилломавирусы, для которых постоянная связь с клеткой организма-хозяина - неотъемлемая часть их жизненного цикла. Вирусы папилломы, как и вирус SV40, относятся к семейству паповавирусов, но они, по-видимому, могут переключаться с инфекции «непродуктивного» типа (лизогенизации) к инфекции «продуктивного» (литического) типа, и наоборот. В первом случае вирус реплицируется синхронно с клеткой, не принося ей вреда, во втором случае он быстро размножается и убивает (лизирует) клетку, высвобождая массу новых вирусных частиц, способных инфицировать другие клетки. Подобно SV40, эти вирусы способны «подчинять себе» клеточную систему синтеза ДНК, а осуществляющие эту функцию вирусные гены могут действовать как онкогены. На рис. 21-20 показано, как, вероятнее всего, вирусы папилломы участвуют в канцерогенезе шейки матки у человека.
21.2.3. Ретровирусы способны случайно захватывать онкогены [15, 18]
В отличие от ДНК-содержащих вирусов большинство ретровирусов (см. разд. 5.5.8 и 13.4.2) относительно безвредны для клетки- хозяина. Зараженная клетка постоянно выделяет новые вирусные частицы, которые отпочковываются от плазматической мембраны, не вызывая неопластической трансформации клетки. Однако изредка может происходить случайное «овладение», захват ретровирусом регуляторного клеточного гена (или его испорченной копии, или фрагмента этого гена), который не используется в жизненном цикле самого вируса, но может кардинально влиять на судьбу клетки-хозяина. В частности, как мы видели в гл. 13 (разд. 13.4.2), ретровирус, подобный вирусу саркомы Рауса, захватившему клеточный онкоген (рис. 21-21), легко обнаруживается по своему доминантному трансформирующему эффекту на инфицированные клетки хозяина, которые начинают бурно размножаться. Онкоген можно идентифицировать и вычленить методами молекулярной генетики, а на его основе синтезировать ДНК-зонды для выявления гомологичных генов в нормальных клетках. Таким способом было выделено более 20 онкогенов, гомологичных протоонкогенам (табл. 21-4), которые имеются в геноме любой нормальной клетки позвоночных (см. разд. 13.4.2). Эти онкогены относятся к нескольким различным семействам (см. разд. 13.4.4), среди них наиболее крупное - семейство протеинкиназных генов, к которому принадлежит и онкоген вируса саркомы Рауса-src (рис. 21-22).
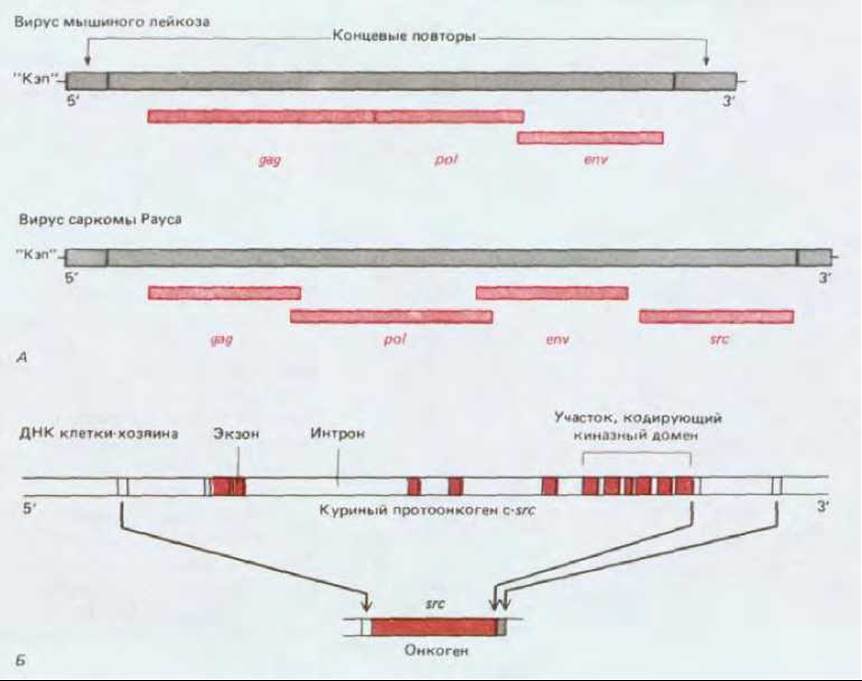
Рис. 21-21. Структура вируса саркомы Рауса. А. Особенности организации генома по сравнению с более типичным ретровирусом (вирус мышиного лейкоза). Вирус саркомы Рауса отличается от других онкогенных ретровирусов в том отношении, что сохранил все три гена, необходимые для его жизненного цикла: gag-кодирующий полибелок, который при «разрезании» дает белки капсида; pol- кодирующий обратную транскриптазу и фермент, участвующий в интеграции хромосомы вируса в геном хозяина; env- кодирующий гликопротеин оболочки. В других онкогенных ретровирусах один или больше вирусных генов отсутствуют, поскольку взамен вирус приобрел трансформирующий онкоген (см. рис. 21-22). В результате трансформирующий вирус может образовывать инфекционные частицы только тогда, когда клетка одновременно заражена недефектным, нетрансформирующим вирусом-хелпером (помощником), который восполняет утерянные функции. Б. Взаимоотношения между онкогеном v-src и клеточным протоонкогеном c-src, из которого произошел онкоген. Присутствующие в гене c-src интроны из гена v-src удалены; кроме того, v-src несет мутации, изменяющие последовательность аминокислот в белке v-src, что делает его гиперактивной нерегулируемой тирозин-специфической протеинкиназой (см. разд. 13.4.5). Исследователи, работающие над проблемой рака, подвергли вирус саркомы Рауса жесткой селекции в отношении его способности трансформировать клетки. Современные штаммы делают это с необычайной быстротой и эффективностью.
Существуют два механизма превращения протоонкогена в онкоген при включении его в ретровирус: изменение последовательности или фрагментация гена, в результате чего на нем синтезируется белок с аномальной активностью, или попадание его (протоонкогена) под контроль мощных вирусных промоторов и энхансеров, что приводит к избыточному накоплению продукта или созданию неподходящих условий для его функционирования; часто происходит и то, и другое. Сходный онкогенный эффект ретровирусы могут оказывать и другим способом, без захвата клеточных генов и переноса их из клетки в клетку: ДНК-копии вирусной РНК могут просто встраиваться в геном клетки рядом с протоонкогенами или даже внутри их. Этот феномен называется вставочным (инсерционным) мутагенезом, а измененный таким образом геном наследуется всеми потомками данной клетки. Вообще, случайное встраивание ДНК-копий вирусной РНК в геном клетки - часть нормального жизненного цикла ретровируса, и если оно происходит в пределах 10 тыс. пар оснований от протоонкогена, то может вызвать аномальную активацию нарушенного встраиванием гена. Вставочный мутагенез дает возможность идентифицировать протоонкогены за счет их близости к встроенному ретровирусу. Выявленные таким способом протоонкогены оказывались теми же, которые обнаруживали и другими методами, но были среди них и новые (табл. 21-5), например, ген int-l, активируемый у мышей, зараженных вирусом опухоли молочных желез (рис. 21-23). Клонирование и секвенирование этого гена выявило его близкое сходство с геном wingless дрозофилы (см. разд. 16.5.9), который, возможно, участвует в межклеточных взаимодействиях, регулирующих сегментацию тела мухи.
Таблица 21-4. Онкогены, первоначально выявленные в составе трансформирующих ретровирусов
|
Онкоген |
Функция протоонкогена (если известна) |
Источник вируса |
Опухоль, вызванная вирусом |
|
abl |
Протеинкиназа (тирозиновая) |
Мышь |
Лейкоз (предшественников В-лимфоцитов) |
|
Кошка |
Саркома |
||
|
akt |
? |
Мышь |
Т-клеточная лимфома |
|
crk |
Активатор тирозин-специфичной протеинкиназы (протеинкиназ) |
Курица |
Саркома |
|
erb-A |
Рецептор гормона щитовидной железы |
Курица |
(совместно с v-erb-B) |
|
erb-B |
Протеинкиназа (тирозиновая) |
Курица |
Эритролейкоз, фибросаркома |
|
Рецептор фактора роста эпидермиса (ФРЭ) |
|||
|
ets |
Ядерный белок |
Курица |
(совместно с V-myb) |
|
fes/fpx |
Протеинкиназа (тирозиновая) |
Кошка/курица |
Саркома |
|
fgr |
Протеинкиназа (тирозиновая) |
Кошка |
Саркома |
|
fms |
Протеинкиназа (тирозиновая) |
Кошка |
Саркома |
|
Рецептор фактора стимуляции колоний макрофагов |
|||
|
fos |
Ядерный фактор транскрипции |
Мышь |
Остеосаркома |
|
jun |
Ядерный белок |
Курица |
Фибросаркома |
|
фактор транскрипции АР-1 |
|||
|
kit |
Протеинкиназа (тирозиновая) |
Кошка |
Саркома |
|
mil/raf |
Протеинкиназа (серин/треониновая) |
Курица/мышь |
Саркома |
|
mos |
Протеинкиназа (серин/треониновая) |
Мышь |
Саркома |
|
myb |
Ядерный белок |
Курица |
Миелобластоз |
|
myc |
Ядерный белок |
Курица |
Саркома миелоцитома карцинома |
|
H-ras |
G-белок |
Крыса |
Саркома эритролейкоз |
|
К-ras |
G-белок |
Крыса |
Саркома эритролейкоз |
|
rel |
Ядерный белок |
Индюк |
Ретикулоэндотелиоз |
|
ros sea |
Протеинкиназа (тирозиновая) |
Курица |
Саркома |
|
Протеинкиназа (тирозиновая) |
Курица |
Саркома, лейкоз |
|
|
sis |
В-цепь тромбоцитарного фактора роста |
Мартышка |
Саркома |
|
ski |
Ядерный белок |
Курица |
Карцинома |
|
src |
Протеинкиназа (тирозиновая) |
Саркома |
|
|
yes |
Протеинкиназа (тирозиновая) |
Курица |
Саркома |
Таблица 21-5. Некоторые онкогены, впервые идентифицированные не по присутствию в трансформирующих ретровирусах
|
Способ идентификации |
Онкогены |
|
Амплификация |
L-myc, N-myc, Gli |
|
Трансфекция |
mas, met, neu, 'N-ras, trk, onc-F, "thy" |
|
Транслокация |
bcl-1, bcl-2, tcl-1, tcl-2, tcl-3a, tcl-3b |
|
Инсерция |
evi-1, int-1, int-2, int-3, int-4, Mlvi-2, Mlvi-3, Pim-1, Flvi-1, Gin-1, fis-1, Ick, dsi-1, fim-1, ahi-1, mis-1, mis-2, mis-3, mis-4, spi |
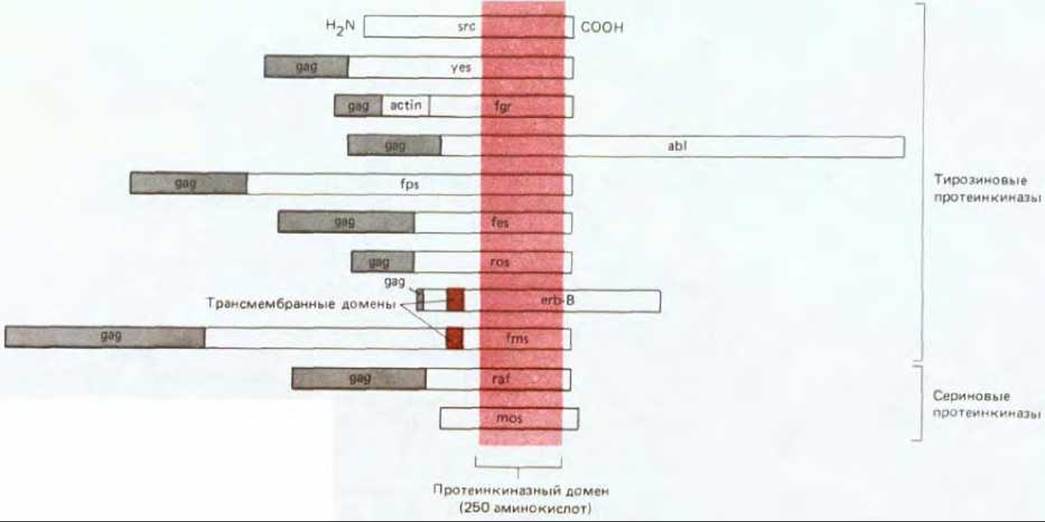
Рис. 21-22. Многие белки, кодируемые онкогенами трансформирующих вирусов, представляют собой протеинкиназы; на рисунке сравниваются (схематично) их первичные структуры. Обратите внимание, что большинство онкогенов несут «осколки» нормальных вирусных генов и продуцируют поэтому «составные» белки, содержащие N-конец вирусного белка gag. (T. Hunter, Sci. Am., 251 (2), 70-79, 1984.)
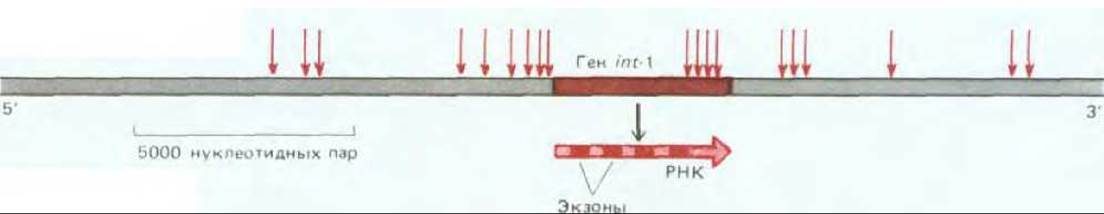
Рис. 21-23. Вставочный мутагенез. В результате вставки активируется ген int-l и возникает опухоль молочной железы у мышей, инфицированных вирусом MMTV (от англ. mouse mammary tumor virus). Участки встраивания вируса, выявленные в 19 различных опухолях, обозначены стрелками. Обратите внимание, что вставки могут активировать транскрипцию гена int-1 на расстоянии более 10 т. п. н. с обеих сторон от гена. Этот эффект обусловлен присутствием мощного энхансера в концевых повторах MMTV.
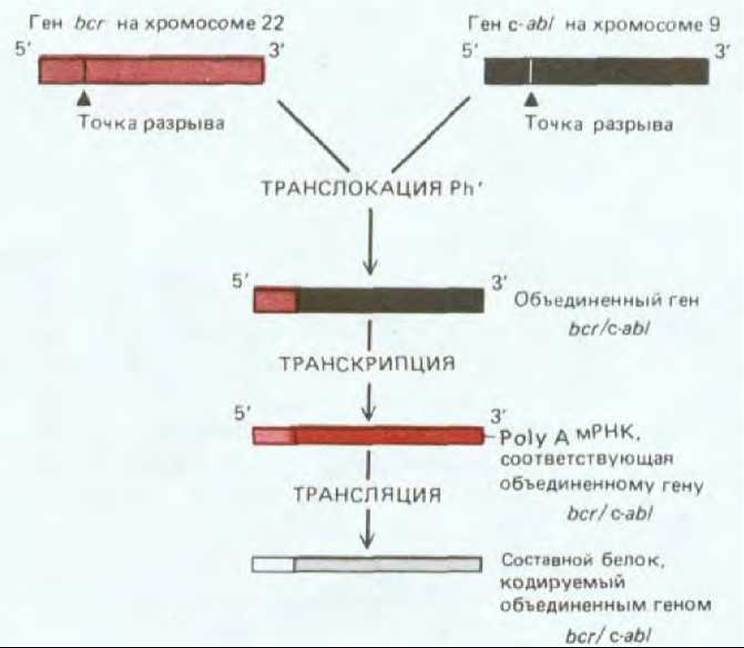
Рис. 21-24. Превращение протоонкогена с-abl в онкоген у пациентов с хроническим миелолейкозом. В результате хромосомной транслокации ген bcr на хромосоме 22 объединяется с геном c-abl из хромосомы 9 (см. рис. 21-4). Образующийся составной белок несет N-концевую последовательность белка bcr и С-концевую последовательность тирозиновой протеинкиназы abl. Abl-киназный домен приобретает аномальную активность, что вызывает избыточную пролиферацию клона гемопоэтических клеток в костном мозге.
21.2.4. Исследование генетической природы рака приводит к одной и той же небольшой группе протоонкогенов [15, 19]
В то время как одни исследователи разрабатывали направление, которое можно назвать «от ретровирусов - к онкогенам», другие использовали более прямой подход и занялись поиском определенных последовательностей ДНК в опухолевых клетках человека, которые могли бы вызвать неконтролируемую пролиферацию при введении в нормальные клетки. Суть метода, разработанного для этой цели, заключалась в трансфекции клеток линии NIH ЗТЗ (одна из неопухолевых линий мышиных клеток ЗТЗ) ДНК, выделенной из клеток опухоли человека (см. разд. 13.4.3) и рис. 13-33). Результат оказался драматический - онкогены были обнаружены во многих линиях раковых клеток человека, и в нескольких случаях эти онкогены оказались мутантными аллелями тех самых протоонкогенов, которые были выявлены при исследованиях с помощью ретровирусов. Так, примерно в каждой четвертой опухоли у человека был найден мутантный представитель генного семейства ras (онкогены, вызывающие саркому у крыс). Таким образом, два независимых пути исследований привели к одной и той же группе генов.
Еще один подход, основанный на кариотипировании опухолевых клеток, дал аналогичный результат. Как мы уже упоминали (разд. 21.1.2), почти у всех больных хроническим миелогенным лейкозом в клетках происходит одна и та же хромосомная транслокация (между хромосомами 9 и 22), а в клетках лимфомы Беркитта обычно наблюдается транслокация между хромосомой 8 и одной из трех хромосом, несущих гены иммуноглобулинов. Было обнаружено, что в обоих типах злокачественных новообразований точка транслокационного разрыва (то место, где часть одной хромосомы соединяется с другой) совпадает с расположением протоонкогенов, уже известных по исследованиям ретровирусов -с-abl при хроническом миелолейкозе и с-тус при лимфоме Беркитта. Аналогичные хромосомные транслокации связаны и с некоторыми другими видами рака. В одних случаях транслокация приводит к превращению протоонкогена в онкоген путем слияния его с другим геном, в результате чего синтезируется измененный белок (рис. 21-24), в других - протоонкоген в результате транслокации попадает в «неестественное» хромосомное окружение, что активирует его транскрипцию и приводит к избыточному синтезу нормального белка.
21.2.5. Существует много способов превращения протоонкогена в онкоген [14, 15, 20]
До настоящего времени выявлено около 60 протоонкогенов (см. табл. 21-4 и 21-5), каждый из которых может быть преобразован в онкоген, играющий решающую роль в возникновении того или иного вида рака. Большинство таких генов обнаруживали неоднократно в самых различных видах опухолей; можно думать поэтому, что большая часть протоонкогенов млекопитающих в настоящее время уже выявлена. За превращение протоонкогена в онкоген могут отвечать разные генетические механизмы (рис. 21-25). Ген может измениться в результате точковой мутации, хромосомной транслокации или вставки мобильного генетического элемента, например ретровируса. Эти изменения могут произойти в участках гена, кодирующих белок (следствием чего является гиперактивный продукт), или в прилежащих регуляторных участках (и тогда возможна простая гиперэкспрессия гена). С другой стороны, гиперэкспрессия может быть и следствием амплификации гена (вероятно, прообразом такого процесса является аномальная репликация хромосом) (рис. 21-26). С некоторыми генами нередко происходят изменения вполне определенного типа (это даже может быть их характерным признаком); точно так же некоторые канцерогены вызывают специфические, «узнаваемые» повреждения. Так, например, гены семейства туе часто экспрессируются в избытке или амплифицируются, а в 90% случаев рака кожи, вызванного у мышей опухолевым инициатором диметилбенз[а] антраценом, наблюдается замена А на Т в одном и том же участке мутантного гена Н-ras.
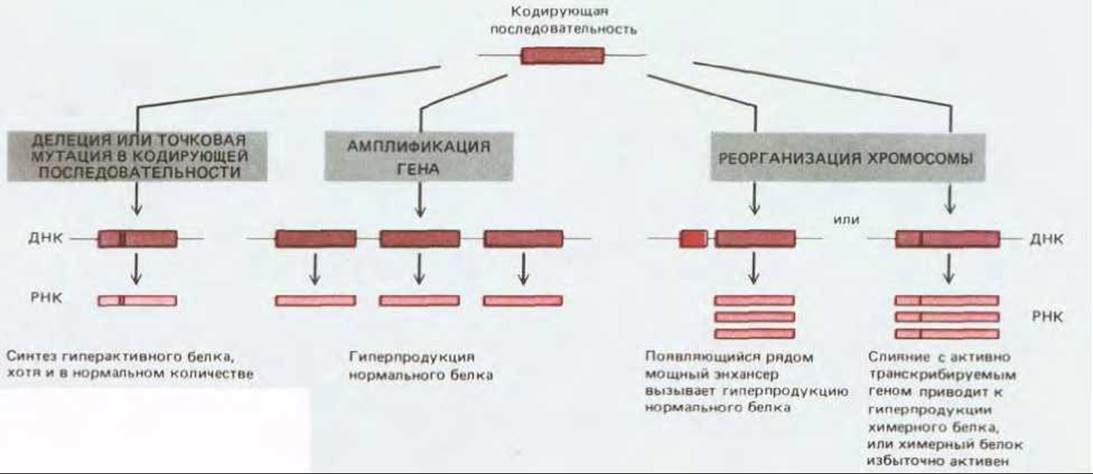
Рис. 21-25. Три пути превращения протоонкогена в онкоген: делеция или точковая мутация (слева), амплификация гена (в центре), хромосомная перестройка (справа). Четвертый механизм (не изображен) включает в себя рекомбинацию между протоонкогеном и ретровирусной ДНК. Конечный эффект подобен тому, который имеет место при реорганизации хромосом: протоонкоген оказывается под контролем вирусного энхансера и/или сливается с активно транскрибируемым вирусным геном.
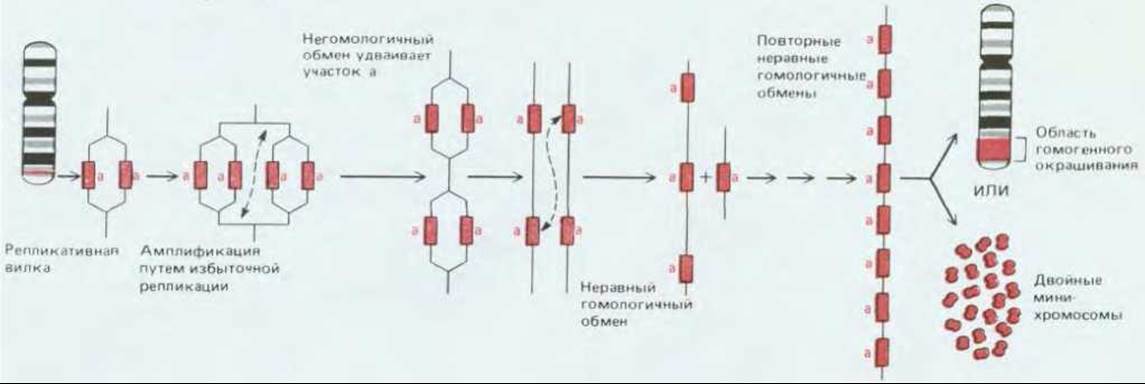
Рис. 21-26. Возможный механизм амплификации гена, приводящей к избыточной продукции белка. Процесс начинается с акта дупликации, в основе которого, по-видимому, лежит незаконная рекомбинация. Изображенная на рисунке схема предполагает, что незаконная рекомбинация может быть следствием дестабилизирующего эффекта избыточной репликации ДНК. Если дупликация гена произошла, неравный обмен сестринских хроматид в результате рекомбинации между одинаковыми копиями генов в ходе репликации ДНК может дополнительно увеличить число копий гена (см. разд. 10.5.2); в результате их количество в хромосоме может достигать десятков и сотен. Многочисленные повторы ДНК делают видимым содержащий их сегмент — он выявляется в хромосоме как область гомогенного окрашивания. Амплифицированный участок может быть также вырезан из своего локуса (видимо, опять же с участием какого-то из рекомбинационных механизмов) и дать начало самостоятельным двойным минихромосомам (см. разд. 21.1.13). Общая длина амплифицированного по такому механизму сегмента ДНК обычно составляет более 100000 нуклеотидных пар.
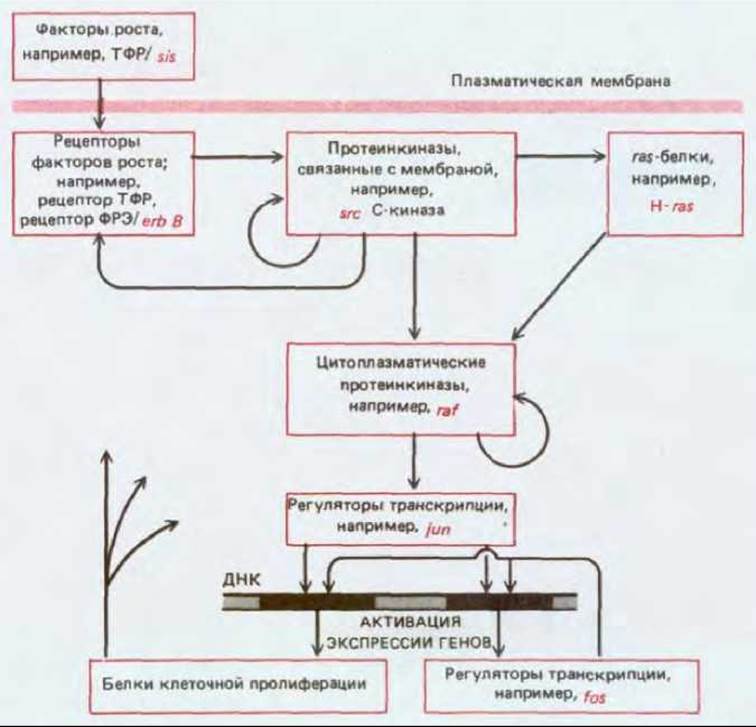
Рис. 21-27. Взаимосвязь между основными классами протоонкогенов во внутриклеточной регуляторной сети, отвечающей за восприятие, передачу и реализацию пролиферативного сигнала. В каждом классе указано по одному характерному представителю. Стрелка от А к Б означает, что в нормальной клетке активация А приводит к активации Б напрямую или опосредованно. В схеме использованы данные, полученные при изучении свойств молекул в бесклеточных системах и, отчасти, при изучении клеток, в которых определенные компоненты были активированы введением онкогена или инактивированы (микроинъекцией соответствующих антител). Существенно, что каждый класс регуляторных молекул представлен многими членами, так что каждая стрелка на схеме «скрывает под собой» множество параллельных стрелок, соединяющих индивидуальные члены одного класса с индивидуальными членами другого. Более того, члены данного класса могут во многих случаях взаимодействовать друг с другом (например, с помощью взаимного фосфорилирования), а также с членами других классов (и предшествующих, и последующих). Наличие множества параллельных путей передачи сигнала, по-видимому, повышает устойчивость клетки к повреждениям, так что единичной онкогенной мутации в норме недостаточно, чтобы сделать клетку опухолевой; однако сложность этой системы и присутствие многочисленных обратных связей делают ее экспериментальное изучение весьма трудной задачей. (Подробности см. в разд. 10.2.8, 12.3.11-12.3.14 и 13.4.4-13.4.6.)
Молекулярные функции протоонкогенов, мутировавших столь разными способами, начинают сейчас проясняться; причем число протоонкогенов, которые прямо вовлечены в какой-либо из путей передачи клеточных сигналов (или имеют к ним отношение, см. гл. 12 и 13, разд. 12.3.10 и 13.4.4), постоянно растет. Продукты многих генов взаимодействуют между собой как компоненты сложной регуляторной сети (рис. 2127). Одни протоонкогены кодируют факторы роста, другие - их рецепторы, а также некоторые протеинкиназы, третьи - G-белки семейства ras или ядерные регуляторные белки. Молекулярные механизмы канцерогенеза обретают, таким образом, «плоть и кровь»: мутации гена с-sis, кодирующего тромбоцитарный фактор роста (ТФР), приводят к его избыточной экспрессии, в результате чего клетки приобретают способность постоянно себя стимулировать; мутации гена с-erbB, кодирующего рецептор к фактору роста эпидермиса (ФРЭ), изменяют его свойства так, как будто он все время активирован своим фактором роста; мутации, усиливающие экспрессию гена с-mус (продукт которого, как полагают, участвует в подготовке ядра к делению), обусловливают способность клетки к неограниченной пролиферации. Тем не менее слишком многое еще остается неясным. Несмотря на богатую информацию о последовательностях ДНК и молекулярной структуре протоонкогенов, онкогенов и их продуктов, мы имеем пока лишь смутное представление о роли большинства из них в физиологии клетки.
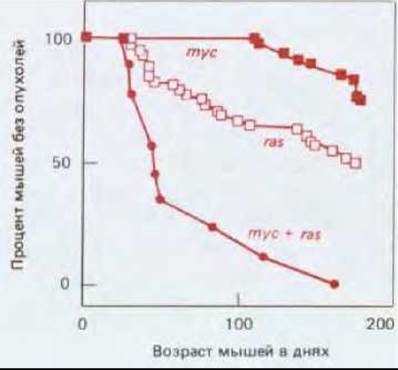
Рис. 21-28.Динамика возникновения опухолей у трансгенных мышей трех типов: несущих онкоген mус, несущих онкоген ras и несущих оба этих онкогена. В ходе эксперимента были использованы две линии трансгенных мышей: одна несла вставленную копию онкогена, полученного слиянием протонкогена с-mус с промотором/энхансером MMTV (этот промотор приводит к избыточной экспрессии находящегося под его контролем гена - в данном случае с-mус - в определенных тканях, например, в молочной железе); другая несла вставленную копию онкогена V-H-ras под контролем того же промотора/энхансера. Опухоли у мышей обеих линий возникали гораздо чаще, чем у нормальных мышей (обычно в молочных и слюнных железах). Мыши, несущие оба онкогена, были получены скрещиванием мышей этих двух линий. Частота возникновения опухолей у таких гибридов была значительно выше, чем суммарная частота для носителей отдельных онкогенов. Несмотря на это, опухоли начинали появляться лишь спустя некоторое время и только у малой доли клеток той ткани, где эти два гена экспрессировались: видимо, для развития рака кроме присутствия двух онкогенов требовалось, чтобы произошло еще какое-то событие (случайного характера). (По Е. Sinn et al., Cell, 40, 465 475, 1987.)
21.2.6. Трансгенные мыши - подходящая тест-система для изучения действия онкогенов [21]
Концепция онкогена содержит в себе противоречие: с одной стороны, в первой части этой главы мы привели множество аргументов в пользу того, что для возникновения рака единичной мутации недостаточно; с другой стороны, онкоген - это доминантный ген, обладающий способностью вызывать неопластическую трансформацию клеток в культуре. Это кажущееся противоречие отражает пропасть между упрощенными моделями рака, наиболее широко используемыми в молекулярно-биологических исследованиях, и сложностью реальной болезни у человека. Стандартный метод идентификации онкогенов выявляет их действие не на нормальные человеческие соматические клетки, а на линию мышиных клеток ЗТЗ; эти клетки уже претерпели мутации в процессе перевода в культуру, что и делает удивительно легкой их трансформацию при единичном дополнительном генетическом изменении. Более того, как указывалось в разд. 21.1.4, мыши в меньшей степени подвержены риску возникновения рака (у них короче продолжительность жизни и меньше общее число клеток), чем человек, и поэтому их клетки могут быть менее надежно защищены от последствий канцерогенных мутаций по сравнению с клетками человека.
Однако даже у мыши единичного онкогена обычно не достаточно для превращения нормальной клетки в раковую. Это отчетливо продемонстрировано на трансгенных мышах. Можно «сшить» онкоген в виде фрагмента ДНК, взятого из вируса или опухолевой клетки, с подходящей промоторной последовательностью ДНК, и ввести полученную молекулу в ядро мышиной яйцеклетки. Такая рекомбинантная молекула ДНК часто встраивается в одну из хромосом, в результате чего получается линия трансгенных мышей, несущих онкоген во всех своих клетках. Этот онкоген может экспрессироваться во многих тканях или же лишь в некоторых «избранных», в соответствии с тканевой специфичностью соединенного с ним промотора. Обычно у мышей, которым ввели таким способом онкогены mус или H-ras, экспрессирующие их ткани резко увеличиваются в объеме, разрастаясь до чрезмерной величины, а отдельные клетки со временем подвергаются дальнейшим изменениям и дают начало опухоли. И все же подавляющее большинство клеток трансгенной мыши, экспрессирующих mус- или H-ras-онкогены, опухолей не образуют, что говорит о недостаточности присутствия одного онкогена для опухолевой трансформации.
Описанный прием был использован для дальнейших исследований. Трансгенных мышей, несущих mус, скрестили с трансгенными мышами, несущими H-ras, и получили потомство, у которого оба онкогена присутствовали одновременно. Такие потомки давали рак с гораздо большей частотой (и гораздо быстрее), чем каждая из родительских линий (рис. 21-28), но опять же он возникал в виде изолированных опухолей, рассеянных среди нераковых клеток. Таким образом, даже с клеткой, экспрессирующей два онкогена, должны произойти какие-то дальнейшие случайные изменения, чтобы она превратилась в опухолевую.
Хотя такой результат является типичным, были обнаружены и исключения, когда клетки какой-нибудь одной ткани у трансгенной мыши, несущей один онкоген, одновременно трансформировались в раковые. Однако в этих случаях онкоген был, по-видимому, «дважды активирован» - благодаря мутации в кодирующем участке и (или) соединению с ненормальной регуляторной последовательностью, что приводило к его избыточной или несоответствующей экспрессии в пораженной ткани. Все это означает, что даже если один онкоген и способен в некоторых случаях вызывать превращение нормальной клетки в опухолевую, единичное генетическое нарушение сделать этого, скорее всего, не может.
21.2.7. Наряду с приобретением онкогенов при раке происходит потеря генов-супрессоров опухолевого роста [22]
Из всех имеющихся ныне фактов создается впечатление, что система контроля и регулирования клеточной пролиферации должна иметь «запас прочности» в том смысле, что никакое единичное генетическое изменение не гибельно для ее нормального функционирования: размножение клеток продолжает оставаться в безопасных границах, даже если любой отдельный компонент системы вышел из строя. Как правило, один онкоген в состоянии оказать свой доминирующий трансформирующий эффект на поведение клетки, лишь тогда, когда система контроля уже серьезно повреждена. Удачи при поиске онкогенов во многом определялись выбором подходящей линии клеток для тестирования.
Значительно труднее оказалось идентифицировать и выделить гены-супрессоры опухолей, т.е. гены, тормозящие в норме избыточную пролиферацию клеток. Тем не менее имеется достаточно доказательств, что потеря таких генов играет определенную роль в патогенезе рака. В частности, в разд. 13.4.3 мы рассказали, что при слиянии культивируемых трансформированных клеток с нетрансформированными возникающие гибриды часто имеют нетрансформированный фенотип. Если же гибрид теряет определенную хромосому, принадлежавшую «нормальному» родителю, его поведение вновь приобретает черты опухолевой клетки. Резонно предположить, что в подобных случаях на утерянной хромосоме находилась единственная клеточная копия специфического гена-супрессора опухолевого роста.
В обычной диплоидной клетке до потери контроля роста и пролиферации должна произойти инактивация или потеря обеих копий опухолесупрессорного гена: мутация, которая участвует в развитии рака за счет инактивации или делеции каких-либо генов, должна быть рецессивной. Напротив, мутация, порождающая онкоген, может влиять на поведение клетки даже в присутствии копии соответствующего протоонкогена. Благодаря этим свойствам дефектные гены-супрессоры опухоли можно распознавать и отличать от онкогенов с помощью простых генетических критериев. Хорошей иллюстрацией служит ретинобластома - редкий вид раковой опухоли, развивающейся в раннем детстве. Опухоль развивается из предшественников нейрональных клеток в незрелой сетчатке. Известны две формы этой опухоли - наследственная и ненаследственная. При наследственной форме обычно возникают множественные опухоли, при этом поражаются оба глаза, а иногда и кости, при ненаследственной ретинообластоме единственная опухоль поражает один глаз. Кроме того, у некоторых больных наследственной формой ретинобластомы выявляется кариотипическая аномалия - исчезновение специфической полосы на 13-й хромосоме. Делеции этого же локуса зафиксированы в клетках опухоли у больных с ненаследственной формой болезни. Эти факты свидетельствуют о том, что причина опухолевого роста заключена скорее в потере гена-супрессора, чем активации онкогена. Имеющиеся данные позволяют сделать вывод, что предрасположенность к раку у больных с наследственной формой обусловлена тем, что в их клетках отсутствует одна из двух нормальных копий некоего гена-супрессора опухолевого роста, названного геном ретинобластомы или RB1. Поэтому теперь достаточно одной соматической мутации, которая испортит оставшуюся «хорошую» копию этого гена в одной из миллиона или более клеток растущей сетчатки, чтобы вызвать рак. У детей без наследственной предрасположенности возникновение ретинобластомы - большая редкость, т. е. для этого необходимо совпадение в одной клетке сетчатки двух соматических мутаций, которые разрушили бы обе копии гена RB1 (рис. 21-29). В настоящее время этот ген клонирован; показано, что он кодирует белок, который в норме экспрессируется как в сетчатке, так и во многих других тканях; он содержит так называемые «цинковые пальцы» (структурные участки, похожие на то, что находят во многих ДНК- связывающих белках, см. разд. 9.1.9), а также связывается с белками - продуктами онкогенов некоторых ДНК-содержащих вирусов. С помощью ДНК-зондов, полученных на основе клонированного RB1, удалось подтвердить, что в опухолевых клетках, в отличие от их нетрансформированных аналогов-соседей, имеются делеции или инактивирующие повреждения гена RBI в обоих - материнском и отцовском - наборах хромосом; потеря нормального гена может осуществляться различными путями (рис. 21-30). Сходные механизмы, но с участием других генов, ответственны за некоторые другие онкологические заболевания раннего детства (например, опухоль Вильмса). Хотя подобные варианты рака редки и в известном смысле являются исключением, накапливается все больше данных в пользу того, что потеря или инактивация супрессорных генов опухолевого роста играет существенную роль в развитии многих раковых заболеваний у лиц зрелого возраста.
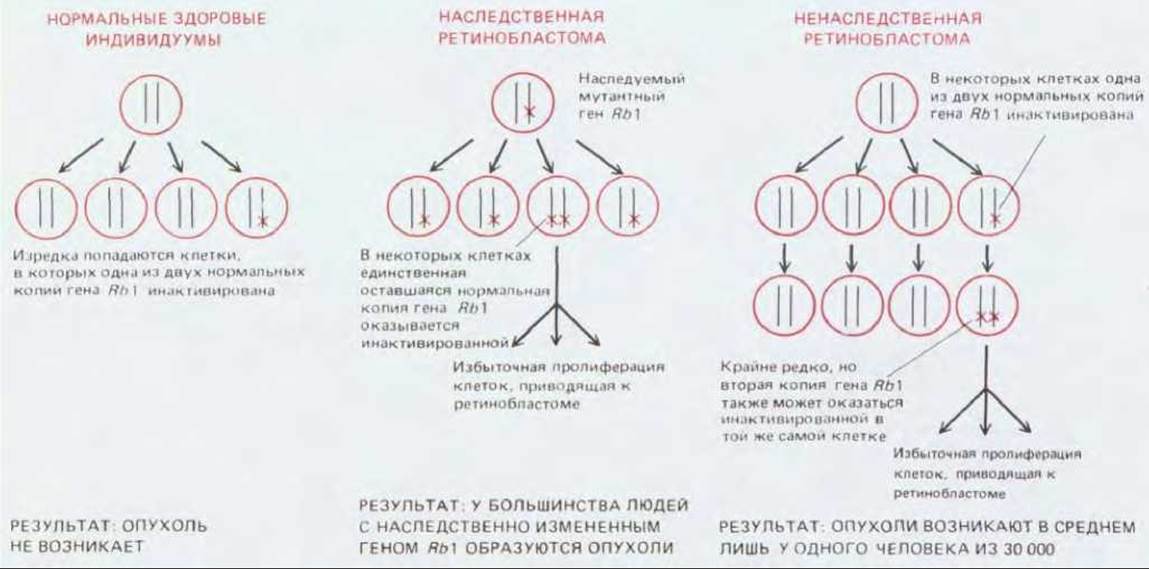
Рис. 21-29. Генетические механизмы возникновения ретинобластомы. При наследственной форме болезни все клетки тела лишены одной из двух (в норме) функциональных копий гена-супрессора опухолевого роста, и при потере или инактивации оставшейся копии в результате соматической мутации в какой-либо из клеток, та может дать начало опухоли. При ненаследственной форме все клетки исходно содержат обе функциональные копии данного гена, и опухоль образуется лишь в том случае, если обе копии утеряны или инактивированы — для этого необходимо совпадение двух соматических мутаций в одной клетке.
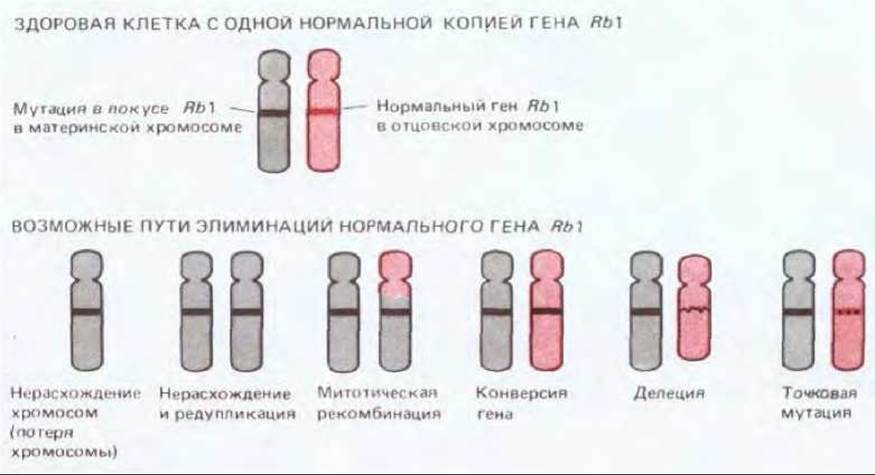
Рис. 21-30. Возможные механизмы, с помощью которых клетка, имеющая дефект в одной из двух копий гена-супрессора опухолевого роста, может лишиться второй, функционально активной копии и сделать тем самым шаг к трансформации. Для анализа опухолевой ДНК и ответа на вопрос, какое именно генетическое событие произошло у данного пациента, весьма эффективны зонды на основе клонированной ДНК в сочетании с исследованием полиморфизма длины рестрикционных фрагментов. Следует отметить, что большинство механизмов приводит к полной потере клеткой функциональной копии (отцовской или материнской) данного гена-супрессора. Оставшийся гомолог изначально дефектен и может оказаться в виде одной или двух генокопий. Подобная редукция до гомозиготности или гемизиготности - характерный признак рака, возникающего в результате потери гена-супрессора опухолевого роста. (По W. К. Cavenee et al., Nature 305, 779-784, 1983.)
21.2.8. Молекулярнобиологический анализ опухолей легких подчеркивает сложность и вариабельность раковых заболеваний у человека [23]
Каждый год в США выявляется около 140 тыс. новых больных раком легкого. До сих пор эффективных методов лечения этой формы рака не существует, и более 90% таких больных умирает в течение года с момента установления диагноза. Для понимания молекулярнобиологических особенностей этого вида злокачественной опухоли были проведены детальные исследования одной из его форм - мелкоклеточного рака легкого, на долю которого приходится 20% всех случаев рака этой локализации. Полагают, что мелкоклеточный рак развивается из нейроэндокринных клеток легкого, секретирующих гастринвысвобождающий пептид (ГВП; его называют также бомбезином из-за сходства с нейропептидом земноводных того же названия) и другие локальные химические медиаторы. Из опухолей различных больных этим видом рака было выделено и проанализировано более сотни культивируемых линий клеток. Эти клетки демонстрируют широкий спектр аномалий, к тому же варьирующих от линии к линии. Выявлены изменения по крайней мере в десяти известных онкогенах и генах-супрессорах опухолевого роста, многочисленные хромосомные делеции и транслокации, а также нарушения в секреции сигнальных молекул, влияющих на клеточную пролиферацию.
Например, многие из этих клеточных линий характеризуются избыточной экспрессией протоонкогена mус или родственных ему генов N- mуc и L-myc. Часто это связано с амплификацией соответствующей области и проявляется в кариотипе как двойные минихромосомы или гомогенно окрашивающиеся хромосомные участки (рис. 21-31, см. также разд. 21.1.13). В этих же клетках гены семейства ras нередко претерпевают специфические точковые мутации. Амплификация гена mус всегда коррелирует с особенно быстрым ростом опухолевых клеток в культуре и с полным подавлением дифференцировки. Больных, имеющих опухоли с амплификацией этого гена, ждет особенно плохой прогноз.
Наряду с другими хромосомными аномалиями, некоторые опухолевые клеточные линии при мелкоклеточном раке легкого имеют значительные хромосомные дефекты в области гена RB1: более чем у половины линий не обнаруживается мРНК этого гена, хотя она синтезируется в нормальных клетках легких и в большинстве раковых клеток другого происхождения. Кроме того, многие опухолевые клеточные линии имеют отчетливые делеции, включающие специфическую полосу 3-й хромосомы, что может указывать на важность потери этого гена для развития рака. С помощью клонированных ДНК-зондов, комплементарных данному участку на 3-й хромосоме, показано, что почти в каждом случае (мелкоклеточного рака и других основных форм рака легкого) одна из двух родительских копий указанного участка на 3-й хромосоме утеряна. В нормальных клетках этих же больных такая делеция не обнаруживается, как и в опухолях других типов у других заболевших. Таким образом, это веский довод в пользу того, что в развитии рака легкого критическим моментом является удаление специфического гена-супрессора опухолевого роста, расположенного в 3-й хромосоме, по одному из механизмов, описанных для ретинобластомы на рис. 21-30. Предполагается, что в типичной раковой легочной клетке одна из двух исходных копий гена-супрессора рака легкого утрачивается за счет делеции сегмента хромосомы, который содержит его, а другая инактивируется вследствие менее заметной (скорее всего, точковой) мутации. По аналогии с ретинобластомой возникает также предположение, что могут быть люди с наследуемым рецессивным дефектом в этом локусе, предрасположенные к раку легкого. Поскольку для возникновения рака легкого, по-видимому, необходимы еще и изменения в других генах (кроме этого для него характерна выраженная зависимость от факторов внешней среды), наследственная предрасположенность в этом случае может быть менее выраженной, чем при ретинобластоме (хотя имеется несколько сообщений о генетической вариабельности чувствительноcти/восприимчивости курильщиков к возникновению рака легкого). Недавно были получены сходные данные о потере гена-супрессора опухолевого роста из другого локуса при другом распространенном раковом заболевании - карциноме толстой кишки; показано, что в этом случае есть редкая наследственная форма, хотя ее взаимосвязь на молекулярном уровне с ненаследуемой формой не выяснена. Можно ожидать, что молекулярнобиологические исследования, подобные только что описанным, помогут выявить людей, с повышенным риском заболеть некоторыми определенными видами рака, и в отношении которых соответствующие превентивные меры могут дать наибольший эффект.

Рис. 21-31. Кариотип опухолевой клетки при мелкоклеточном раке легкого. Стрелками указаны двойные минихромосомы, содержащие амплифицированный ген mус. (С любезного разрешения F. Whang-Peng и F. D. Minna.)
Другой клинически важный результат, полученный при молекулярнобиологическом исследовании мелкоклеточного рака легкого, касается бомбезина (ГВП). Этот пептид действует как фактор роста, стимулируя пролиферацию опухолевых клеточных линий. Некоторые линии клеток (но не все) оказались способными не только отвечать на этот фактор, но и секретировать его, создавая аутокринную петлю, посредством которой клетки сами стимулируют себя к делению. В культуре пролиферацию таких клеток удается затормозить с помощью антител, связывающих пептид и блокирующих тем самым стимуляцию. Можно надеяться, что в будущем станет возможным останавливать таким способом опухолевый рост у пациентов с этой разновидностью рака легкого.
21.2.9. Каждый случай рака представляет собой самостоятельный природный эксперимент в клеточной эволюции
Всякий успех в лечении зависит прежде всего от верного диагноза. Если мы не можем точно идентифицировать болезнь, то мы не сумеем ни обнаружить ее причины, ни предсказать ее исход, ни подобрать подходящее для данного больного лечение. Как мы могли убедиться на примере мелкоклеточного рака легкого, традиционная классификация онкологических заболеваний неточна, одна из общепринятых в ней категорий при внимательном рассмотрении превращается в гетерогенную группу болезней, каждая из которых имеет свой характерный набор генетических повреждений. Молекулярная биология начинает создавать инструменты, с помощью которых можно точно узнать, какие гены амплифицированы, какие делетированы и какие подверглись мутациям в опухолевых клетках любого конкретного больного. Информация такого рода столь же важна для лечения и предотвращения рака, как идентификация возбудителя при инфекционных заболеваниях.
Открытие онкогенов и, совсем недавно, генов-супрессоров опухолевого роста ознаменовало конец эры блужданий во тьме в поисках биохимической основы рака. Однако предстоит еще пройти путь от упрощенных лабораторных моделей, сделавших эти триумфальные успехи возможными, к пониманию реальной сложности раковых заболеваний человека. Пока мы далеки от успеха, прогресс до обидного невелик и в отношении разработки эффективного рационального лечения. Мы знаем последовательности ДНК многих онкогенов и протоонкогенов, но в то же время точные физиологические функции нам известны лишь для некоторых из них. Необходимо более глубокое понимание того, как эти и другие молекулы взаимодействуют между собой, управляя поведением отдельной клетки; нужно хорошо разбираться в популяционных механизмах, которые определяют возникновение раковых клеток через мутации и естественный отбор.
Каждый онкологический больной - это жертва неудачного эксперимента в клеточной эволюции, поставленного самой природой. Сложность (и запутанность) самого феномена рака обусловливается сложностью и богатством эволюционных возможностей, и в то же время именно эволюционные принципы позволяют решать проблему рака. Итак, мы возвращаемся к идее, с которой была начата эта книга: все в биологии имеет смысл лишь в свете эволюции.
Заключение
Размножение нормальных клеток регулируется ингибирующими и стимулирующими молекулами, которые являются соответственно продуктами генов-супрессоров опухолевого роста и протоонкогенов. Проявление раковых свойств у клетки может быть результатом как потери или инактивации обеих клеточных копий гена-супрессора, так и амплификации или гиперактивации одной из двух копий протоонкогена. Наследуемые нарушения пролиферативного контроля могут быть вызваны также внедрением в клетку чужеродного вирусного генетического материала. Ретровирусы могут сами становиться онкогенными, «захватывая» копию клеточного протоонкогена клетки-хозяина и превращая его в онкоген; они могут также создавать онкоген в клетке, действуя как инсерционный мутаген и внедряясь в ее геном рядом с протоонкогеном. Хотя полагают, что большинство онкологических заболеваний у человека вызывается не вирусами, обнаруживаемые в опухолевой ДНК мутации часто затрагивают те же протоонкогены, что и найденные при изучении ретро-вирусов. Способы превращения протоонкогенов в онкогены в опухолях у человека включают точковые мутации, амплификацию генов, а также хромосомные транслокации, которые могут привести к нарушению контроля экспрессии этого протоонкогена или к его соединению с другим геном с последующим синтезом нового белка. Подобно этому, гены-супрессоры опухолевого роста могут быть функционально утеряны в результате мутаций самого разного характера; люди, унаследовавшие от родителей делецию или дефектную копию одного из таких генов, могут проявить выраженную предрасположенность к определенному типу рака, что демонстрирует пример с ретинобластомой. Молекулярнобиологический анализ опухолевых клеток от больных, страдающих одной из наиболее распространенных форм рака, выявил сложный и неоднородный спектр генетических повреждений, включая и активацию онкогенов и потерю генов-супрессоров опухолевого роста. Эти данные являются отражением случайного характера эволюционного процесса, в ходе которого возникает рак, и говорят о том, что каждая злокачественная опухоль, с молекулярной точки зрения уникальна.
Литература
Общая
Cairns J. Cancer: Science and Society. San Francisco, Freeman, 1978.
Cancer Biology: Readings from Scientific American. New York, W. H. Freeman, 1986.
Darnell J.E., Lodish H.F., Baltimore D. Molecular Cell Biology, pp. 1035-1080. New York, W. H. Freeman/Scientific American Books, 1986.
De Vita V.T., Hellman S., Rosenberg S.A., eds. Cancer: Principles and Practice of Oncology, 2nd ed. Philadelphia, Lippincott, 1985.
Farmer P. В., Walker J. M., eds. The Molecular Basis of Cancer. New York. Wiley, 1985.
Franks L. M., Teich N., eds. Introduction to the Cellular and Molecular Biology of Cancer. Oxford, U.K., Oxford University Press, 1986.
Prescott D.M., Flexer A. S. Cancer: The Misguided Cell, 2nd ed. Sunderland, M. A., Sinauer, 1986.
Watson A. M., Hopkins N. H., Roberts J. W., Steitz J. A., Weiner A. M. Molecular Biology of the Gene, 4th ed., pp. 962-1096. Menlo Park, CA, Benjamin-Cummings, 1987.
Цитированная
1. Cairns J. Mutation selection, and the natural history of cancer. Nature, 255, 197-200, 1975.
Nowell P. C. The clonal evolution of tumor cell populations. Science, 194, 23-28, 1976.
2. Doll R., Peto R. Epidemiology of cancer. In Oxford Textbook of Medicine (D.J. Weatherall, J.G.G. Ledingham, D.A. Warrell, eds.), 2nd ed., pp.
4.95-4.123. Oxford U.K., Oxford University Press, 1987.
Parkin D. M., Laara E., Muir C. S. Estimates of the worldwide frequency of sixteen major cancers in 1980. Int. J. Cancer, 41, 184-197, 1988. Ritchie A. C. The classification, morphology, and behaviour of tumours. In General Pathology (H. Florey, ed.), 4th ed., pp. 668-719. London, Lloyd-Luke, 1970.
Robbins S. L., Cotran R. S., Kumar V. Pathologic Basis of Disease, 3rd ed., pp. 214-272. Philadelphia, Saunders, 1984.
3. Fearon E. R., Hamilton R., Vogelstein B. Clonal analysis of human colorectal tumors. Science, 238, 193-197, 1987.
Fialcow P. J. Clonal origin of human tumors. Biochem. Biophys. Acta, 458, 283-321, 1976.
Groffen J.J., et al. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell, 36, 93-99, 1984. Nowell P. C., Hungerforcl D. A. A minute chromosome in human granulocytic leukemia. Science, 132, 1497, 1960.
4. Ames В., Durston W.E., Yamasaki E., Lee F.D. Carcinogens are mutagens: a simple test system combining liver homogenates for activation and
bacteria for detection. Proc. Natl. Acad. Sci. USA, 70, 2281-2285, 1073.
Ashby J., Tennant R. W. Chemical structure, Salmonella mutagenicity and extent of carcinogenecity as indicators of genotoxic carcinogenesis among 222 chemicals tested in rodents by the U.S. NCI/NTP. Mutation Res., 204, 17-115, 1988.
Case R. M. P. Tumours of the urinary tract as an occupational disease in several industries. Ann. R. Coll. Surg. Engl., 39, 213-235, 1966.
Doll R. Strategy for detection of cancer hazards to man. Nature, 265, 589-597, 1977.
Hay A. How to identify a carcinogen. Nature, 332, 782-783, 1988.
Walcer J. M. Testing for carcinogens. In The Molecular Basis of Cancer (P. B. Farmer, J. M. Walcer, eds.), pp. 181-200. New York, Wiley, 1985.
5. Armitage P., Doll R. The age distribution of cancer and a multi-stage theory of carcinogenesis. Br. J. Cancer, 8, 1-12, 1954.
Peto R. Cancer epidemiology, multistage models and shortterm mutagenicity tests. In Origins of Human Cancer (H. H. Hiatt, J. D. Watson, J. A. Winsten, eds.), pp. 1403-1428. Cold Spring Harbor, NY, Cold Spring Harbor Laboratory, 1977.
6. Cairns J. Cancer. Science and Society, pp. 144 156. San Francisco, Freeman, 1978.
Campion M.J., McCance D.J., Cuzick ., Singer A. Progressive potential of mild cervical atypia: persrective, cytological, colposcopic, and virological study. Lancet, 2, 237-240, 1986.
Farber E., Cameron C. The sequential analysis of cancer development. Adv. Cancer Res., 32, 125-225, 1980.
Foulds L. The natural history of cancer. J. Chronic. Dis., 8, 2-37, 1958.
Mclndoe W.A., McLean M. R., Jones R. W., Millins P.R. The invasive potential of carcinoma in situ of the cervix. Obstet. Gynecol., 64, 451464, 1984.
7. Albanes D., Winick M. Are cell number and cell proliferation risk factors for cancer? J. Natl. Cancer. Inst., 80, 772 775, 1988.
Schinipper L. E. Clinical implications of tumor-cell heterogeneity. N. Engl. J. Med., 314, 1423-1431, 1986.
8. Berenblum I. A speculative review: the probable nature of promoting action and its significance in the understanding of the mechanism of carcinogenesis. Cancer Res., 14, 471-477, 1954.
Berenblum I., Shibik P. The role of croton oil applications associated with a single painting of a carcinogen in tumor induction in the mouse's skin. Br. J. Cancer, 1, 379-383, 1947.
Dolberg D.S., Holligsworth R., Hertle M., BisselM.J. Wounding and its role in RSV-mediated tumor formation. Science, 230, 676 678, 1985. Lamph W. W., Wamsley P., Sassone-Corsi P., Verma I. M. Induction of proto-oncogene JUN/AP-1 by serum and TPA. Nature 334, 629-631, 1988.
Reddy A.L., Fialcow P.J. Influence of dose of initiator on two-stage skin carcinogenesis in BALB/c mice with cellular mosaicism. Carcinogenesis, 9, 751-754, 1988.
9. Cairns J. The treatment of diseases and the war against cancer. Sci. Am., 253(5), 51-59, 1985.
Cohen L.A. Diet and cancer. Sci. Am., 257(5), 42-48, 1987. Doll R., Peto R. The Causes of Cancer. New York, Oxford University Press, 1981. Enstrom J. E. Cancer and mortality among active Mormones. Cancer, 42, 1943-1951, 1978.
Pike M.C., Ross R.K. Breast cancer. Br. Med. Bull., 40, 351-354, 1984.
10. Fentiman I. The local treatment of cancer. In Introduction to the Cellular and Molecular Biology of Cancer (L. M. Franks, N. Teich, eds.), pp. 350-362. Oxford, U.K., Oxford University Press, 1986.
Malpas J. Chemotherapy. In Introduction to the Cellular and Molecular Biology of Cancer (L. M. Franks, N. Teich, ed.), pp. 363-377. Oxford, U. K., Oxford University Press, 1986.
11. Sachs L. Growth, Differentiation, and the reversal of malignancy. Sci. Am., 254(1), 40-47, 1986.
12. Fidler L, Hart I. Biological diversity in metastatic neoplasms: origins and implications. Science, 217, 998-1003, 1982.
Liotta L. A. Tumor invasion and metastases - role of the extracellular matrix: Rhoads Memorial Award lecture. Cancer. Res., 46, 1-7, 1986. McCarthy J.B., Skubitz A.P.N., Palm S.L., Furcht L.T. Metastasis inhibition of different tumor types by purified laminin fragments and a heparin-binding fragment of fibronectin. J. Natl. Cancer Inst., 80, 108-116, 1988.
Nicholson G. Cancer metastasis. Sci. Am., 240(3), 66-76, 1979.
Schirrmacher V. Cancer metastasis: experimental approaches, theoretical concepts, and impacts for treatment strategies. Adv. Cancer Res., 43, 173, 1985.
13. German J., ed. Chromosome Mutation and Neoplasia. New York, Liss, 1983.
Hanawalt P., Sarasin A. Cancer-prone hereditary diseases with DNA processing abnormalities. Trends Genet., 2, 124-129, 1986.
Lindahl T. Regulation and deficiencies in DNA repair. Br. J. Cancer, 56, 91095, 1987.
Nowell P. C. Mechanisms of tumor progression. Cancer Res., 46, 2203-2207, 1986.
14. Alt F. W., Kellems R. E., Bertino J. R., Schimke R. T. Selective multiplication of dihydrofolate reductase genes in methotrexate-resistant variants of cultured murine cells. J. Biol. Chem., 253, 1357-1370, 1978.
Gerlach J. H., et al. Homology between P-glycoprotein and a bacterial haemolysin transport protein suggests a model for multidrug resistance. Nature, 324, 485-489, 1986.
Roninson l. В., Abelson H. Т., Houseman D. E., Howell N., Varshavsky A. Amplification of specific DNA sequences correlates with multi-drug resistance in Chinese hamster cells. Nature, 309, 626-628, 1984.
Schimke R. T. Gene amplification in cultured cells. J. Biol. Chem., 263, 5989-5992, 1988.
15. Klein G. The approaching era of the tumor suppressor genes., Science, 238, 1539-1545, 1987.
Watson J.D., Hopkins N.H., Roberts J. W., Steitz J.A., Weiner A.M. Molecular Biology of the Gene, 4th ed., Chapters 26-27. Menlo Park, CA, Benjamin-Cummings, 1987.
Wyke J. A. Viruses and Cancer. In Introduction to the Cellular and Molecular Biology of Cancer (L. M. Franks, N. Teich, eds.), pp. 176-199. Oxford, U.K., Oxford University Press, 1986.
16. Kripke M.L. Immunoregulation of carcinogenesis: past, present and future. J. Natl. Cancer Inst., 80, 722-727, 1988.
17. Laso P. A. Human papillomaviruses in oncogenesis. Bioessays, 9, 158-162, 1988. Steinherg B. M., Brandsma J. L., Taichman L. B. Cancer Cells 5/Papillomaviruses. Cold Spring Harbor, NY, Cold Spring Harbor Laboratory, 1987.
18. Bishop J.M. Oncogenes. Sci. Am., 246(3), 80-92, 1982.
Rijsewijk F., et al. The Drosophila homolog of the mouse mammary oncogene int-1 is identical to the segment polarity gene wingless. Cell, 50, 649-657, 1987.
Varmus H. E. The molecular genetics of cellular oncogenes. Annu. Rev. Genet., 18, 553-612, 1984.
19. Bishop J.M. The molecular genetics of cancer. Science, 235, 305-311, 1987.
Croce C. M., Klein G. Chromosome translocations and human cancer. Sci. Am., 252(3), 44-50, 1985. Weinberg R.A. A molecular basis of cancer. Sci. Am., 249(5), 126-142, 1983.
20. Bradshaw R. A., Prentis S. Oncogenes and Growth Factors. Amsterdam, Elsevier, 1987. (Подборка статей из журналов Trends Biochem. Sci., Trends Genet, и Immunol. Today.)
Hunter T. The proteins of oncogenes. Sci. Am., 251(2), 70-79, 1984.
Kahn P., Graf Т., eds. Oncogenes and Growth Control. Berlin, Springer, 1986. (Прекрасная коллекция коротких обзоров.)
Quintanilla M., Brown К., Ramsden M., Balmain A. Carcinogen-specific mutation and amplification of Ha-ras during mouse skin carcinogenesis. Nature, 322, 78-80, 1986.
Smith M. R., DeGudicibus S.J., Stacey D. W. Requirement for c-ras proteins during viral oncogene transformation. Nature, 320, 540 543, 1986. (В статье иллюстрируются возможности использования антител для анализа взаимодействия протоонкогенов в регуляции клеточных функций.)
21. Hanahan D. Dissecting multistep tumorigenesis in mice. Annu. Rev. Genet. 22 [в печати], 1988.
Land H., Parada L.F., Weinberg R.A. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature, 304, 596-602, 1983.
Quaife C.J., Pinkert C.A., Ornitz D.N., Palmiter R.D., Brinster R.L. Pancreatic neoplasia induced by ras expression in acinar cells of transgenic mice. Cell, 48, 1023-1034, 1987.
Sinn E., et al. Coexpression of MMTV/v-Ha-ras and MMTV/c-mvc genes transgenic mice: synergistic action of oncogenes in vivo., Cell, 49, 465-475, 1987.
22. Hansen M. F., Cavenee W. K. Retinoblastoma and the progression of tumor genetics. Trends Genet., 4, 125-129, 1988.
Harris H. The genetic analysis of malignancy. J. Cell Sci. (suppl.), 4, 431-444, 1986.
Knudson A. G. Hereditary cancer, oncogenes, and antioncogenes. Cancer Res., 45, 1437-1443, 1985. Lee E. Y.-H. P., et al. Inactivation of the retinoblastoma susceptibility gene in human breast cancer. Science, 241, 218-221, 1988.
Weinberg R.A. Finding the anti-oncogene. Sci. Am., 259(3), 34 41, 1988.
23. Bodmer W. F., et al. Localization of the gene for familial adenomatous polyposis on chromosome 5. Nature, 328, 614-627, 1987.
Harbour J. W., et al. Abnormalities in structure and expression of the human retinoblastoma gene in SCLC. Science, 241, 353-357, 1988. (SCLC-мелкоклеточный рак легкого.)
Kok К., et al. Deletion of a DNA sequence at the chromosomal region 3p21 in all major types of lung cancer. Nature, 330, 561-578, 1987. Leppert M., et al. The gene for familial polyposis coli maps to the long arm of chromosome 5. Science, 258, 1411-1413, 1987.
Minna J. D., et al. Molecular genetic analysis reveals chromosomal deletion, gene amplification, and autocrine growth factor production in the pathogenesis of human lung cancer. Cold Spring Harbor Symp. Quant. Biol., 51, 843-853, 1986.