ИММУНОЛОГИЯ - Ройт А. - Мир 2000
Глава 21. Первичная иммунологическая недостаточность
■ При нарушениях иммунного ответа повышается чувствительность организма к пиогенным (гноеродным) бактериям. Подобные нарушения могут возникнуть вследствие недостаточности функции В-клеток, как это имеет место при сцепленной с Х-хромосомой агаммаглобулинемии. В других случаях причина состоит в том, что В-клетки не получают соответствующих сигналов от Т-клеток. С этим связано возникновение таких расстройств, как синдром IgM-гипергаммаглобулинемии, общий вариабельный иммунодефицит и транзиторная гипогаммаглобулинемия детского возраста.
■ При недостаточности клеточного иммунитета организм подвержен оппортунистическим инфекциям. Такой тип иммунодефицита обусловлен нарушением функций Т-клеток, как это имеет местопри тяжелом комбинированном иммунодефиците (ТКИД), недостаточности молекул МНС класса II, атаксии-телеангиэктазии, синдромах Вискотта-Олдрича и Ди Джорджи.
■ Наследственная патология системы комплемента обнаруживается при ряде клинических синдромов. Наиболее часто ее дефект состоит в недостаточности ингибитора С1, клинически проявляющейся как ангионевротический отек.
■ При наследственной недостаточности терминальных компонентов комплемента (С5, С6, С7 и С8) и белков альтернативного пути активации комплемента (фактора Н, фактора I и пропердина) чрезвычайно повышена чувствительность к инфекциям, вызываемым двумя видами Neisseria - N. gonorrhoeae и N. meningitidis.
■ Нарушения механизма восстановления молекулярного кислорода в фагоцитах, а именно сборки молекулы NADP·H-оксидазы и образования активных метаболитов кислорода, обладающих бактерицидными свойствами, служат причиной развития хронического гранулематоза. Длительное присутствие бактериальных продуктов в фагоцитах ведет к образованию либо абсцессов, либо гранулем, в зависимости от вида возбудителя.
■ Недостаточность адгезии лейкоцитов ассоциирована с персистирующим лейкоцитозом, поскольку лейкоциты крови, несущие дефектные молекулы интегринов, не способны проникать черезсосудистый эндотелий в ткани.
Иммунодефицитные состояния возникают в результате выпадения или недостаточности функции одного или нескольких элементов иммунной системы. Причинами заболеваний, обусловленных специфической иммунной недостаточностью, служат нарушения функций Т- или В-лимфоцитов — основы приобретенного иммунитета. Неспецифические иммунодефициты связаны с нарушениями в таких элементах иммунной системы, как комплемент и фагоциты, действующих при иммунном ответе неспецифично. Первичные иммунодефицитные состояния обусловлены внутренними дефектами клеток иммунной системы и большей частью генетически детерминированы.
При иммунодефицитном состоянии наблюдается повышенная чувствительность к инфекциям. Наиболее часто возникающие у таких больных инфекции можно разделить на две категории. При нарушениях, связанных с иммуноглобулинами, компонентами комплемента и фагоцитарной активностью, резко возрастает восприимчивость к повторным инфекциям, вызываемым бактериями, которые обладают капсулой, — Haemophilus influenzae, Streptococcus pneumoniae и Staphylococcus aureus. Эти бактерии называют пиогенными, или гноеродными, поскольку они вызывают гнойное воспаление. В случаях нарушений в системе клеточного иммунитета, т. е. функциях Т-клеток, повышается чувствительность к микроорганизмам, широко распространенным во внешней среде и в норме безвредным - у здоровых людей к ним быстро развивается резистентность, но у больных с недостаточностью Т-клеточной функции они способны вызывать генерализованные и даже летальные инфекции. Это так называемые оппортунистические инфекции; их возбудителями могут быть различные микроорганизмы, от дрожжей до обычных вирусов, таких как вирус ветряной оспы.
В-КЛЕТОЧНАЯ НЕДОСТАТОЧНОСТЬ
Больные с общими дефектами В-клеточной функции (рис. 21.1) подвержены рецидивирующим пиогенным инфекциям, таким как пневмония, воспаление среднего уха и синусит. При отсутствии лечения повторные пневмонии могут вызвать тяжелое обструктивное заболевание органов дыхания (бронхоэктаз) вследствие разрушения эластических тканей бронхиальной стенки.

Рис. 21.1. Недостаточность В-лимфоцитов может проявляться в различных формах, например, как задержка продукции нормальных иммуноглобулинов, дефицит одного из изотипов Ig или Х-сцепленная агаммаглобулинемия, при которой в организме отсутствуют В-клетки, а сыворотка не содержит иммуноглобулинов.
При сцепленной с Х-хромосомой агаммаглобулинемии (Х-АГ) нарушено созревание В-клеток на ранней стадии их развития
Этот синдром иммунодефицита, описанный в 1952 г., стаз первым детально изученным расстройством такого рода. Он встречается только у мальчиков и характеризуется отсутствием или резким снижением числа В-клеток в крови или лимфоидных тканях, из-за которого лимфатические узлы у больных очень мелкие, а миндалины отсутствуют. В сыворотке крови обычно отсутствуют IgA, IgM, IgD или IgE, а количество IgG пониженное (< 100 мг/100 мл). В первые 6-12 мес жизни ребенок защищен от инфекций за счет IgG, полученных при трансплацентарном переносе от матери в период внутриутробного развития. Но поскольку со временем эти IgG катаболизируются, у страдающих данным нарушением мальчиков возникают рецидивирующие гнойные инфекции. Здоровье таких детей можно поддерживать, только вводя им внутривенно большие дозы гамма-глобулина.
Ген Х-АГ картирован в длинном плече Х-хромосомы (рис. 21.2). Здесь же локализованы многие гены, ответственные за другие наследственные синдромы иммунодефицита: обнаружение дефектов этих генов весьма существенно дли пренатальной диагностики. Недавно проведенные исследования позволили идентифицировать дефектный ген при Х-АГ. Им оказался ген цитоплазматической тирозинкиназы В-клеток (btk), принадлежащий к семейству онкогена src. Роль этого гена в созревании В-клеток еще нe выяснена, однако очевидно, что он имеет решающее значение для развития В-лимфоцитов. Костный мозг у больных Х-АГ содержит нормальное число пре-В-клеток, однако вследствие мутаций гена btk они не могут превратиться в зрелые В-лимфоциты (рис. 21.3).

Рис. 21.2. Гены, с которыми связаны многие синдромы иммунодефицита, локализованы в Х-хромосоме. При всех этих синдромах обнаружены генетические дефекты. (По Schwaber J., Rosen F.S., 1990. X-chromosome linked immunodeficiency. Immunodefic. Rev. 2: 233-51.)
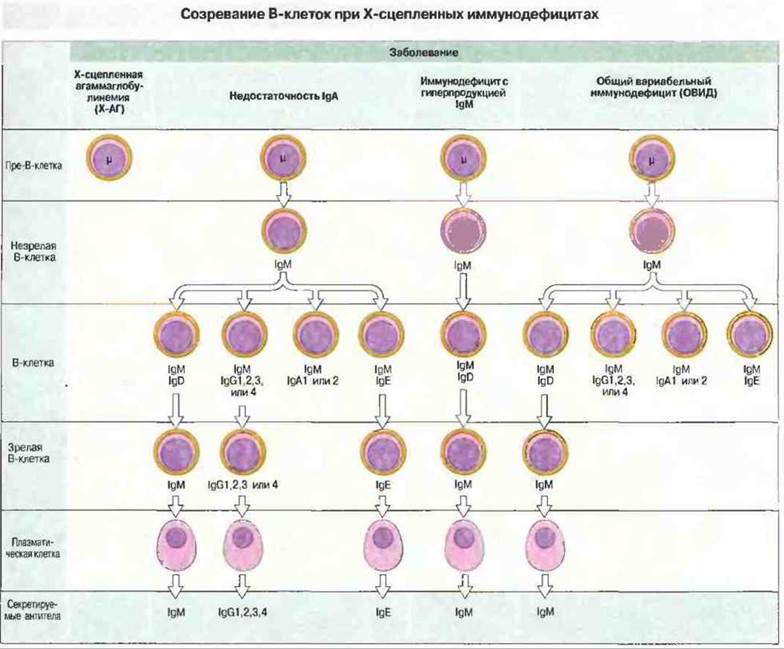
Рис. 21.3. У мальчиков, больных Х-АГ, отсутствуют В-клетки и сывороточные иммуноглобулины, за исключением небольшого количества IgG материнского происхождения. При IgA-недостаточности В-клетки, несущие IgA, а в некоторых случаях также IgG2 и IgG4, неспособны дифференцироваться в плазматические клетки. У лиц, страдающих иммунодефицитом с повышенным содержанием IgM, отсутствуют IgG и IgA. При ОВИД В-клетки большинства изотипов не могут дифференцироваться в плазматические.
При дефиците иммуноглобулинов IgA и IgG не происходит конечной дифференцировки В-клеток
Наиболее часто встречающаяся форма иммунодефицита - это отсутствие или резкое снижение уровня IgA в сыворотке крови. У европеоидов частота этой формы составляет 1 на 700, однако у представителей других рас она не обнаруживается или встречается очень редко. Лица с недостаточностью IgA подвержены заболеваниям, в развитии которых играют роль иммунные комплексы (гиперчувствительность III типа). Примерно у 20 % больных с дефицитом IgA отсутствуют также IgG2 и IgG4, что сильно повышает чувствительность к гноеродным микроорганизмам. Антитела против капсульных полисахаридов гноеродных бактерий у человека относятся преимущественно к подклассу IgG2, поэтому сам по себе дефицит IgG2 также ведет к возникновению повторных гнойных инфекций. При недостаточности только IgG4 больные, кроме того, особо подвержены рецидивирующим инфекциям (причина этого неясна). Дефицит иммуноглобулинов указанного класса и подкласса возникает в результате нарушения конечного этапа дифференцировки В-клеток (рис. 21.3).
При иммунодефиците с гиперпродукцией IgM (ГИГМ) не происходит переключения изотипа
Особая форма иммунологической недостаточности — это дефицит IgG и IgA в сочетании с образованием большего количества поликлональных IgM (>200 мг/100 мл). Такое состояние сопровождается повышенной чувствительностью к гнойным инфекциям и требует лечения высокими дозами вводимого внутривенно гамма-глобулина. При нем наблюдается продукция IgM-аутоантител к нейтрофилам, тромбоцитам и другим элементам крови, а также к тканевым антигенам, т. е. состояние иммунодефицитности у таких больных осложнено аутоиммунными процессами. Некоторые ткани, особенно желудочно-кишечного тракта, инфильтрированы IgM-продуцирующими клетками (рис. 21.4). При ГИГМ в В-клетках не происходит переключения с синтеза IgM на образование IgG, IgA и IgE, что обычно имеет место при В-клеточной дифферениировке. В нормальных условиях такое переключение обусловлено двумя факторами: связыванием ИЛ-4 с рецептором к ИЛ-4 на поверхности В-клеток и взаимодействием молекул CD40 на поверхности В-клеток с лигандом, CD40L, на активированных Т-клетках. В 70 % случаев ГИГМ наследуется по сцепленному с Х-хромосомой рецессивному типу; она обусловлена мутациями в гене лиганда CD40, поскольку этот ген картирован точно в том же участке длинного плеча X-хромосомы, что и ген, ответственный за ГИГМ.
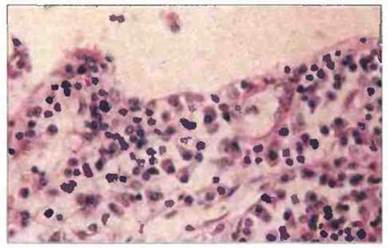
Рис. 21.4. Желчный пузырь больного с синдромом IgМ-гипергаммаглобулинемии. Подслизистая основа инфильтрирована клетками с окрашенной в розовый цвет цитоплазмой и эксцентрично расположенным ядром. Эти клетки синтезируют и секретируют IgM.
Общий вариабельный иммунодефицит (ОВИД) связан с нарушением передачи сигналов от Т- к В-клеткам
При ОВИД у больных в возрасте от 10 до 30 лет или старше развивается агаммаглобулинемия. Оба пола поражаются с одинаковой частотой, и в общем причина заболевания неизвестна, хотя оно может возникнуть после вирусной инфекции, в частности вызванной вирусом Эпштейна-Барр (ВЭБ). Как и при Х-АГ, больные с ОВИД чрезвычайно чувствительны к гноеродным микроорганизмам и кишечному паразиту Giardia lamblia (рис. 21.5), вызывающему тяжелую диарею. У большинства больных (80 %) с ОВИД В-клетки не функционируют надлежащим образом и являются незрелыми. Они не имеют дефекта, но просто не получают необходимых для активации сигналов от Т-клеток. Очевидно, причина этого кроется в каком-то дефекте Т-лимфоцитов, однако его природа при ОВИД не определена достаточно точно. Для предупреждения повторных гнойных инфекций больным необходимо в течение всей жизни вводить внутривенно гамма-глобулин. У многих из них возникают аутоиммунные заболевания, особенно часто злокачественная анемия, этиологический фактор которой неизвестен. ОВИД не является наследственным заболеванием, однако он ассоциирован с МНС-гаплотипами HLA-B8 и HLA-DR3.
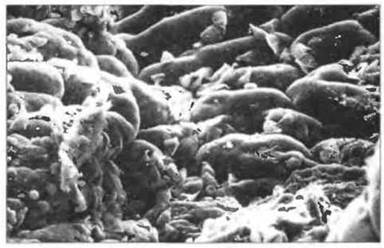
Рис. 21.5. Giardia lamblia. Множество лямблий - клеток паразитического простейшего рода Giardia - на поверхности слизистой оболочки тощей кишки у больного с ОВИД.
Транзиторная гипогаммаглобулинемия детского возраста характеризуется задержкой образования IgG
Выше уже отмечено (см. рис. 21.3), что противоннфекционная зашита у детей в раннем постнатальном периоде обусловлена наличием IgG, полученных от матери до рождения. Этот IgGкатаболизируется с периодом полураспада около 30 сут. В обычных условиях у детей начиная с возраста 3 мес вырабатываются собственные IgG, хотя продукция антител против капсульных полисахаридов бактерий в лучшем случае начинается только на втором году жизни. У некоторых детей начало нормального синтеза IgG наблюдается не раньше трехлетнего возраста, и до этого они особо подвержены гнойным инфекциям. В-клетки таких детей не имеют дефектов, однако, по- видимому, не получают от Т-клеток CD4+достаточной помощи, необходимой для продукции антител.