ИММУНОЛОГИЯ - Ройт А. - Мир 2000
Глава 4. Комплемент
БИОЛОГИЧЕСКИЕ ЭФФЕКТЫ КОМПЛЕМЕНТА
Биологические активности системы комплемента можно подразделить на полезные для организма-хозяина и вредные.
Основные полезные эффекты комплемента:
✵ содействие в уничтожении микроорганизмов;
✵ интенсивное удаление иммунных комплексов;
✵ индукция и усиление гуморального иммунного ответа.
Система комплемента может вызывать повреждение клеток и тканей собственного организма в следующих случаях;
✵ если происходит ее генерализованная массированная активация, например при септицемии, вызванной грамотрицательными бактериями;
✵ если ее активация происходит в очаге тканевого некроза, в частности при инфаркте миокарда;
✵ если активация происходит при аутоиммунной реакции в тканях.
Комплемент способствует уничтожению микроорганизмов. Усиление ликвидации микробов достигается несколькими путями, включая:
✵ образование анафилатоксинов, которые повышают проницаемость стенок сосудов, облегчая тем самым поступление в очаг инфекции других защитных факторов воспалительной реакции;
✵ опсонизация микробов для усиления фагоцитоза;
✵ внедрение лизирующего мембрану комплекса в мембрану микробных клеток.
Анафилатоксины - сильные индукторы воспаления. Активация системы комплемента приводит к образованию анафилатоксинов С3а и С5а, физиологическая роль которых состоит в привлечении клеток воспалительного экссудата в очаг воспаления, а также в активации их эффекторных механизмов.
Системное введение С5а или генерализованная внутрисосудистая активация комплемента (например, при сепсисе, вызванном грамотрицательными бактериями), может привести к сердечно-сосудистому коллапсу и бронхоспазму — т. е. к состоянию, напоминающему анафилаксию (отсюда название анафилатоксины).
Активности С5а. С5а служит сильным активатором всех типов клеток миелоидного ряда (рис. 4.18). Этот анафилатоксин вызывает хемокинез и хемотаксис нейтрофилов, их дегрануляцию, а также вспышку клеточного дыхания с образованием кислородных радикалов. Кроме того, С5а вызывает метаболизирование арахидоновой кислоты, входящей в состав мембран, с образованием простагландинов и эйкозаноидов. При этом возрастает также поверхностная экспрессия молекул межклеточной адгезии, что способствует прилипанию клеток к сосудистому эндотелию (рис. 4.18). У моноцитов и макрофагов С5а вызывает аналогичные реакции и, кроме того, секрецию ИЛ-1 и ИЛ-6, а у базофилов и тучных клеток - дегрануляцию с высвобождением гистамина и других вазоактивных медиаторов.
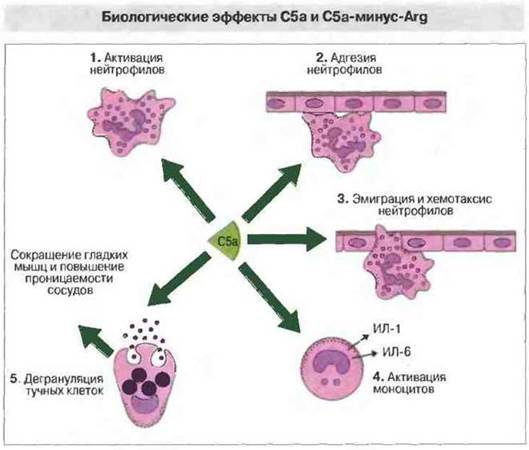
Рис. 4.18. Анафилатоксин С5а вызывает 1) активацию нейтрофилов, 2) повышенную экспрессию ими молекул межклеточной адгезии, 3) эмиграцию нейтрофилов и хемотаксис, 4) активацию моноцитов и 5) дегрануляцию тучных клеток, в результате которой происходит сокращение гладкой мускулатуры и повышение проницаемости сосудов.
Активируя эти клетки, С5а опосредованно влияет на кровеносные сосуды, повышая их проницаемость, и на гладкую мускулатуру, вызывая ее сокращение. Кроме того, С5а может действовать синергично с другими медиаторами воспаления, например, вместе с ИФγ или эндотоксином стимулировать секрецию ИЛ-1 моноцитами.
Время полужизни С5а. Присутствие С5а в кровотоке весьма кратковременно, как и следует ожидать для столь мощного медиатора воспаления. Содержащийся в крови фермент карбоксипептидаза N отщепляет от С5а С-концевой остаток аргинина, в результате чего все биологические активности этого эффектора, за исключением хемотаксической, существенно ослабевают. Затем происходит связывание его рецептором для С5а, интернализация и быстрое внутриклеточное расщепление протеазами на неактивные фрагменты.
Активность С3а. По сравнению с С5а этот субкомпонент комплемента обладает гораздо меньшей активностью и связывается с иным клеточным рецептором. Он вызывает слабую агрегацию нейтрофилов и вспышку клеточного дыхания, но в противоположность С5а не обладает хемотаксической активностью.
Отметим, что образование анафилатоксинов происходит в результате активации не только комплемента, но и других ферментных систем, которые непосредственно расщепляют С3, С4 и С5. К таким ферментам относятся плазмин, каликреин, тканевые и лейкоцитарные (лизосомные) протеазы (в частности, эластаза нейтрофилов), а также протеолитические ферменты микробного происхождения, например гингипаин-1 из бактерии Porphyromonas gingivalis, которая встречается при патологии периодонта.
Фиксированные С3b и С4b действуют как опсонины, усиливая фагоцитоз. Ковазентно связываясь с поверхностью бактерий и иммунными комплексами, С3b и С4b делают их лигандами для рецепторов комплемента на фагоцитарных клетках. Тем самым они обеспечивают очистку крови от бактерий и иммунных комплексов. Связывание С3b и С4b с рецепторами комплемента на поверхности нейтрофилов, моноцитов и макрофагов может вызывать, кроме стимуляции фагоцитоза, экзоцитоз гранул, содержащих протеолитические ферменты, и образование свободных кислородных радикалов в результате вспышки клеточного дыхания (рис. 4.19).
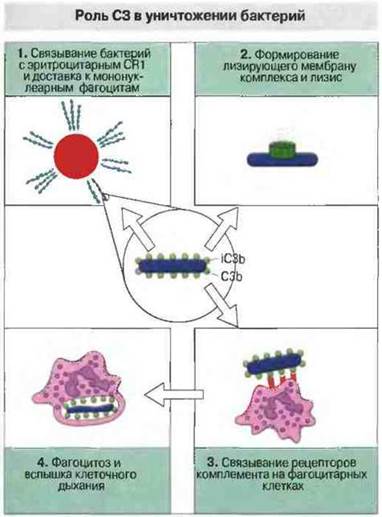
Рис. 4.19. Компонент С3, связанный с бактериальной клеткой в виде С3b или iC3b, 1) взаимодействует с CR1 эритроцитов, на которых бактерии транспортируются кровотоком, 2) служит «причалом» для лизирующего мембрану комплекса на поверхности бактериальных клеток, 3) «сшивает» рецепторы комплемента на фагоцитах, 4) активирует фагоциты, стимулируя фагоцитоз, вспышку клеточного дыхания и бактерицидную активность.
Недостаточность комплемента ассоциирована с подверженностью инфекционным заболеваниям. Физиологическая роль комплемента в опсонизации и бактериолизе становится совершенно ясной, если проанализировать две формы его наследственной недостаточности. Недостаточность компонентов классического пути и С3 и недостаточность семейства рецепторов CR3/CR4/LFA-1 ассоциированы с частым возникновением инфекций, вызываемых гноеродными бактериями. Тот факт, что дефицит опсонинов либо рецепторов приводит к одинаковым последствиям, убедительно свидетельствует о важной роли комплемента в уничтожении этих бактерий путем фагоцитоза и внутриклеточного разрушения.
В отличие от этого недостаточность компонентов Л МК ассоциирована почти исключительно с повышенной восприимчивостью к заражению Neisseria meningitidis. Можно предполагать, что устойчивость организма в отношении этой бактерии, которая способна выживать внутри фагоцитов, основана на комплемент-зависимом бактериолизе в плазме крови.
Менее важен, по-видимому, комплемент для противовирусной зашиты, в которой решающая роль принадлежит Т-клеткам. Недостаточность комплемента обычно не сопровождается повышенной восприимчивостью к вирусным инфекциям.
Лизирующий мембрану комплекс принимает также участие в воспалительной реакции. Согласно традиционному представлению, ЛМК уничтожает любую клетку, образуя поры в мембране и вызывая ее лизис. Однако не так давно было установлено, что содержащие ядро клетки, например клетки иммунной системы организма, относительно устойчивы к литическому действию ЛМК; отчасти это обусловлено присутствием на мембране регуляторных молекул типа CD59, но, кроме того, и способностью этих клеток устранять путем эндоцитоза или экзоцитоза те участки своей плазматической мембраны, в которые проник ЛМК. Даже в случае сублетального воздействия ЛМК вызванное им изменение структуры мембранного бислоя может стимулировать клетки иммунной системы (в зависимости от их тканевого происхождения) к высвобождению и метаболизированию арахидоновой кислоты, усилению окислительного метаболизма, дегрануляции или секреции цитокинов. Эти реакции, возможно, важны для усиления воспаления в участках активации комплемента.
Патогенные микроорганизмы противодействуют эффектам комплемента
Взаимодействие между системой комплемента и микробами можно рассматривать как фактор продолжающейся эволюционной межвидовой борьбы. По мере развития системы комплемента, вероятно под давлением отбора, связанного главным образом с инфекционными заболеваниями, у микробов в свою очередь появились механизмы выхода из-под удара комплемента и даже «использования» этой системы для развития инфекции. Фактически, патогенные микробы патогенны именно благодаря своей способности обходить в известной мере механизмы зашиты организма от инфекции.
Грамотрицательные бактерии экспонируют связывающие С3b и ЛМК структуры, на которых бактериолитическая активность комплемента лишена эффективности. Наружный слой клеточной стенки большинства грамотрицательных бактерий содержит липополисахарид (ЛПС) с длинными О-специфическими боковыми полисахаридными цепями, выступающими из мембраны наружу. Они эффективно активируют комплемент, но локализуют ковалентное связывание С3 и фиксацию ЛМК на таком удалении от цитоплазматической мембраны бактериальной клетки, при котором опсонизация и лизис невозможны. В подобных случаях в качестве фактора приобретенного иммунитета могут функционировать только бактерицидные антитела. Они активируют комплемент в непосредственной близости к тем участкам бактериальной поверхности, где его опсонизирующий и литический эффекты могут реализоваться.
Некоторые бактерии имеют наружный покров, устойчивый к опсонизации. Ряд микроорганизмов устойчив к действию комплемента за счет присутствия на их поверхности молекул, препятствующих альтернативной активации комплемента и усилению фиксации С3. Например, штаммы патогенных грамположительных бактерий отличаются от своих непатогенных аналогов наличием богатой сиаловыми кислотами капсулы, на которой С3b связывает фактор Н, а не фактор В, в результате чего подвергается расщеплению.
Микробы могут экспрессировать молекулы, подавляющие активацию комплемента. Другая стратегия обхода микробами действия комплемента - это экспрессия ингибиторов, подобных тем, которыми обладает организм-хозяин. Известны присутствующие на поверхности бактериальных клеток молекулы с Fc-рецепторными свойствами, например стафилококковый белок А и Fc-рецептор, имеющийся у многих герпесвирусов. Недавно обнаружена также экспрессия рецептора (гликопротеин-С) для комплемента вирусом простого герпеса. Грибы Candida albicansэкспрессируют молекулы, подобные CR2 и CR3 и имеющие даже антигенное сходство с CR3 человека. Все эти молекулы способны защитить микроорганизмы от обычных последствий связывания антител и комплемента. Так, IgG или С3, связавшись с рецепторами на поверхности микробов, могут утратить способность к взаимодействию с Fc-рецепторами на фагоцитарных клетках. Еще один стратегический путь заключается в экспрессии регуляторных молекул, подавляющих активацию комплемента. Так, например, трипаносомы образуют ФУД- и СD59-подобные молекулы, тогда как шистосомы просто адсорбируют ФУД организма-хозяина, достигая той же цели.
Некоторые вирусы используют систему комплемента для усиления своего патогенного действия
Наиболее важная стадия в патогенезе вирусных инфекций — это проникновение возбудителя в клетки организма-хозяина. Как установлено, некоторые вирусы используют связанные с клеточными мембранами компоненты комплемента в качестве рецепторов для усиления проникновения в клетку. Так, вирус Эпштейна-Барр использует CR2, вирус кори — мембранный кофакторный белок (МКБ, CD46), а ряд эховирусов — фактор, ускоряющий диссоциацию С3-конвертазы (ФУД, CD55).
Проникновению ряда других вирусов в клетки может способствовать их связывание с антителами и С3b в жидкой фазе. В частности, в результате взаимодействия с антителами усиливается поглощение клетками флавивирусов (в том числе вируса денге) при участии макрофагальных Fc-рецепторов, а связывание С3 с вирусными частицами способствует поглощению вируса Западного Нила (также флавивируса) при участии CR3.
Комплементу принадлежит важная вспомогательная роль в индукции иммунного ответа
Система комплемента облегчает контакт и взаимодействие антигенпрезентирующих клеток и В-клеток с антигеном (рис. 4.20). Например, от комплемента зависит необходимая для формирования В-клеток памяти локализация иммунных комплексов в центрах размножения внутри лимфоузлов.
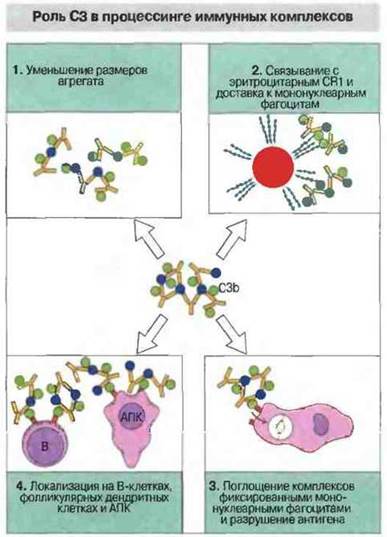
Рис. 4.20. Компонент С3 связывается с иммунными комплексами и благодаря этому 1) уменьшает размеры иммунных агрегатов решетчатой структуры, 2) опосредует связывание циркулирующих иммунных комплексов с CR1 на эритроцитах, которые транспортируют эти комплексы в кровотоке, 3) способствует поглощению иммунных комплексов фиксированными мононуклеарными фагоцитами и тем самым разрушению антигена и 4) способствует локализации антигена в виде иммунных комплексов на В-лимфоцитах и антигенпрезентирующих клетках, в том числе на специализированных фолликулярных (дендритных) клетках лимфоузлов.
На В-клетках и АПК выявлены следующие рецепторы комплемента:
✵ В-клетки: CR1, связывающий С3b и iC3b, а также CR2, связывающий iC3b и C3dg;
✵ моноциты и макрофаги: CR1 и CR3;
✵ фолликулярные дендритные клетки (единственный тип клеток, обладающий всеми тремя рецепторами): CR1, CR2 и CR3.
Лица с наследственным дефицитом С3 страдают лишь умеренным нарушением продукции антител. Однако у морских свинок, дефицитных по С2, С3 или С4, наблюдается заметное угнетение первичного и вторичного иммунных ответов на малые дозы Т-зависимых антигенов. Эти факты свидетельствуют о вспомогательной (но не решающей) роли комплемента в эффективной индукции образования антител.
Комплемент участвует в процессинге иммунных комплексов
В 1940-х гг. Гейдельбергером было установлено, что комплемент препятствует формированию решетчатой структуры преципитирующих комплексов антиген—антитела. На структуру и размеры иммунных комплексов влияют многие факторы, включая следующие:
✵ концентрация реагентов (антител и антигена);
✵ аффинность антител к гомологичному антигену;
✵ валентность как антител, так и антигена (чем выше валентность, тем крупнее образующиеся комплексы).
Активация комплемента по классическому пути подавляет образование преципитатов иммунных комплексов в плазме крови. Подобным же образом, активация по альтернативному пути может вызвать растворение иммунных комплексов, уже образовавших преципитаты в плазме, а также в тканях. Растворение происходит в результате ковалентного включения СЗ в решетчатую структуру иммунного преципитата: С3 разрушает связь антител с эпитопами антигена, ограничивая тем самым возможность образования крупных агрегатов (см. гл. 25).
Активация комплемента иммунными комплексами в норме физиологически полезна, так как связанные с С3 комплексы эффективно удаляются из тканей и кровотока моноцитами и прочими фагоцитарными клетками (рис. 4.20). Однако в некоторых случаях интенсивное образование иммунных комплексов продолжается хронически, и тогда активация ими комплемента имеет вредные последствия; в частности это происходит при подостром бактериальном эндокардите и системной красной волчанке.
Комплемент способствует развитию некоторых заболеваний
Системная активация комплемента приводит к образованию больших количеств анафилатоксинов. В определенных условиях активация комплемента in vivo играет вредную, а не полезную роль (рис. 4.21). Например, шок при бактериемии, вызванной грамотрицательными бактериями, отчасти обусловлен системной активацией комплемента эндотоксином. Возникающие при этом в больших количествах С3а и С5а вызывают активацию и дегрануляцию нейтрофилов, базофилов и тучных клеток. Внутрисосудистая агрегация нейтрофилов приводит к диссеминированному свертыванию крови и задержке образовавшихся микроэмболов в капиллярах легких, где продукты лейкоцитарного происхождения (включая эластазу и свободные радикалы) могут вызвать синдром «шокового легкого». Он характеризуется интерстициальным отеком легкого вследствие повреждения мелких сосудов, образованием нейтрофильного экссудата в альвеолах и артериальной гипоксемией.
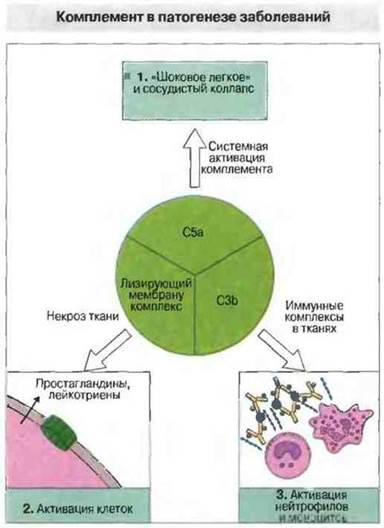
Рис. 4.21. Активация комплемента может вызвать патологические реакции в результате 1) системного образования анафилатоксинов (например, при септицемии, вызванной грамотрицательными бактериями), 2) внедрения лизирующего мембрану комплекса в мембраны собственных клеток организма (при этом происходит активация клеток и высвобождение метаболитов арахидоновой кислоты, входящей в состав мембран) и 3) фиксации С3 (привлекающего и активирующего тканевые и циркулирующие лейкоциты) на иммунных комплексах, локализованных в тканях.
Искусственное кровообращение через аппараты сердце-легкие или купрофановые диализаторы может стать причиной экстракорпоральной активации комплемента, которая сопровождается временной лейкопенией, примерно такой же, как при агрегации нейтрофилов в легких.
Тканевой некроз активирует комплемент. Повреждение ткани вследствие ишемического некроза способно вызвать локальную активацию комплемента и интенсивную фиксацию ЛМК на клеточной мембране. О возможной патофизиологической роли активации комплемента в этом случае свидетельствуют данные экспериментального моделирования инфаркта миокарда, при котором снижение концентрации комплемента уменьшает масштабы повреждения ткани. Подобный же эффект, как установлено недавно, вызывает введение растворимого рекомбинантного CR1.
Активация комплемента вследствие образования иммунных комплексов in vivo - возможная причина повреждения тканей. Активация комплемента имеет существенное значение в патогенезе тканевой деструкции при заболеваниях, обусловленных образованием иммунных комплексов. Формирование таких комплексов возможно в тканях, например в почечных клубочках при нефропатии, вызванной образованием аутоантител к гломерулярной базальной мембране, или на концевых пластинках двигательных нейронов при злокачественной миастении с образованием аутоантител к холинорецепторам (см. гл. 24). В других случаях циркулирующие иммунные комплексы могут отлагаться в стенках кровеносных сосудов (см. гл. 25). Например, при бактериальном эндокардите инфицированный сердечный клапан представляет собой источник образования иммунных комплексов, которые оседают в почках или других участках микрососудистого русла.
При болезнях иммунных комплексов комплемент провоцирует воспаление главным образом двумя следующими путями:
✵ с С3b и С4b, фиксированными на иммунных комплексах, связываются лейкоциты, активируемые и привлекаемые в места отложения этих комплексов образовавшимися здесь анафилатоксинами; так начинается повреждение тканей при синдроме Гудпасчера и для подавления воспалительной реакции на экспериментальных моделях этого заболевания достаточно уменьшить содержание в крови комплемента или нейтрофилов;
✵ ЛМК (лизирующий мембрану комплекс) повреждает клеточную мембрану и стимулирует при этом образование простагландинов из арахидоновой кислоты. Этим обусловлено повреждение тканей при мембранозном нефрите, который в эксперименте удается вызвать антителами к субэпителиальным антигенам. Воспалительную реакцию в этом случае не подавляет устранение нейтрофилов, однако она почти полностью отсутствует у животных, дефицитных по С5. Базальная мембрана, вероятно, служит физическим барьером на пути миграции нейтрофилов, поэтому наблюдаемая высокая протеинурия обусловлена только фиксацией лизирующего мембрану комплекса.
Вопросы для размышления
■ Каков механизм защиты тканей организма от повреждения собственной системой комплемента?
■ С чего начинается активация комплемента?
■ Как может повлиять наследственная недостаточность фактора Н на систему комплемента ребенка? Какие заболевания возможны у таких детей?
■ В чем состоят эффекторные механизмы системы комплемента?
■ Что представляет собой внутренняя тиоэфирная связь в структуре С3 и С4? Каково ее значение для системы комплемента?
■ Чем обусловлена та избирательность, благодаря которой активация комплемента происходит на поверхности чужеродных клеток, а не собственных клеток организма?
■ Как взаимодействуют между собой система комплемента и микробы?
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
Ahearn J.M., Fischer М.В., Croix D. et al. 1996. Disruption of the Cr2 locus results in a reduction in B-1a cells and in an impaired В cell response to T- dependent antigen. Immunity 4: 251-262.
Aulak K.S., Donaldson V.H., Coutinho M. et al. 1993. C1-inhibitor: structure/function and biologic role. Behring Inst. Mitt. 93: 204-213.
Bkakdi S., Tranum Jensen J. 1991. Complement lysis: a hole is a hole. Immunol. Today 12: 318-320.
Campbell R.D., Law S.K.A., Reid K.B.M. et al. 1988. Structure, organization and regulation of the complement genes. Annu Rev. Immunol. 6: 161-195.
Colten H.R., Rosen F.S. 1992. Complement deficiencies. Annu. Rev. Immunol. 10: 809-834.
Cooper N.R. 1985. The classical complement pathway: activation and regulation of the first complement component. Adv. Immunol. 37: 151-216.
Davies K.A., Schifferli J.A., Walport M.J. 1994. Complement deficiency and immune complex disease. Springer Semin. Immunopathol. 15: 397-416.
Dodds A.W., Ren X.D., Willis A.C. et al. 1996. The reaction mechanism of the internal thloester in the human complement component C4. Nature 379: 177-179.
Esser A.F. 1991. Big MAC attack: complement proteins cause leaky-patches. Immunol. Today 12: 316-318.
Farries T.C., Atkinson J.P. 1991. Evolution of the complement system. Immunol. Today 12: 295-300.
Fearon D.T., Locksley R.M. 1996. The instructive role of innate immunity in the acquired immune response. Science 272: 50-53.
Frank M.M. 1992. The mechanism by which microorganism avoid complement attack. Curr. Opion. Immunol. 4: 14-19.
Frank M.M., Fries L.F. 1991. The role of complement in inflammation and phagocytosis. Immunol. Today 12: 322-326.
Gerard C., Gerard N.P. 1994. C5a anaphylatoxin and its seven transmembrane-segment receptor. Annu. Rev. Immunol. 12: 775-808.
Holmskov U., Mathotra R., Sim R.B. et al. 1994. Collectins: collagenous C-type lectins of the innate immune defense system. Immunol. Today 14: 67-74.
Hourcade D., Holers V.M., Atkinson J.P. 1989. The regulators of complement activation (RCA) gene cluster. Adv. Immunol. 45: 381-416.
Joiner K.A. 1988. Complement evasion by bacteria and parasites. Annu. Rev. Microbiol. 42: 201-230.
Kinoshita T, Inoue N., Takeda J. 1996. Role of phos- phatidylinositol-linked proteins in paroxysmal nocturnal hemoglobinuria pathogenesis. Annu. Rev. Med. 47: 1-10.
Lachmann P.J., Walport M.J. 1987. Deficiency of the effector mechanisms of the immune response and autoimmunity. In: Whelan J. (ed.). Autoimmunity and Autoimmune Diseases. Chichester: Wiley, 149-171.
Liszewski M.K., Parries T.C., Lublin D.M. et al. 1996. Control of the complement system. Adv. Immunol. 61:201-283.
Moffitt M.C., Frank M.M. 1994. Complement resistance in microbes. Springer Semin. Immunopathol. 15: 327-344.
Morgan B.P. 1995. Complement regulatory molecules: application to therapy and transplantation. Immunol. Today 16:257-259.
Morgan B.P., Meri S. 1994. Membrane proteins that protect against complement lysis. Springer Semin. Immunopathol. 15: 369-396.
Morgan B.P., Walport M.J. 1991. Complement deficiency and disease. Immunol. Today 12: 301-306.
Muller-Eberhard H.J. 1986. The membrane attack complex of complement. Annu. Rev. Immunol. 4:503-528.
Muller-Eberhard H.J., Schreiber R.D. 1980. Molecular biology and chemistry of the alternative pathway of complement. Adv. Immunol. 29: 1-53.
Reid K.B.M., Day A J. 1989. Structure-function relationships of the complement components. Immunol. Today 10: 177-180.
Reid K.B.M., Porter R.R. 1981. The proteolytic activation systems of complement. Annu. Rev. Biochem. 50. 433-464.
Reid K.B.M., Turner M.W. 1994. Mammalian lectins in activation and clearance mechanisms involving the complement system. Springer Semin. Immunopathol. 15: 307-326.
Ross G.D. (ed.) 1986. Immunobiology of the Complement System. New York. Academic Press.
Smith G.L. 1994. Virus strategies for evasion of the host response to infection. Trends Microbiol. 2: 81-88.
Walport M.J. 1993. Inherited complement deficiency - clues to the physiological activity of complement in vivo. Q.J. Med. 86: 355-358.
Walport M.J., Lachmann P.J. 1993. Complement. In: Lachmann P.J., Peters D.K., Rosen F.S., Walport M.J. (eds.). Clinical Aspects of Immunology, 5th edn. Oxford: Blackwell Scientific Press., 347-375.