Практическая химия белка - А. Дарбре 1989
Пептидное картирование белков
Картирование пептидов на практике
Двумерное картирование пептидов
Очищенные белки подвергают гидролизу трипсином (иногда а-химотрипсином) в растворе, в суспензии [26] или в геле [19]. При небольшом количестве исходного материала необходимо проводить предварительно радиоактивное мечение [6, 19, 20]. Немеченые пептиды могут быть обработаны после разделения флуоресцентными реагентами, причем образующиеся производные в некоторых случаях по чувствительности обнаружения не уступают радиоактивной метке [21, 55, 60, 70].
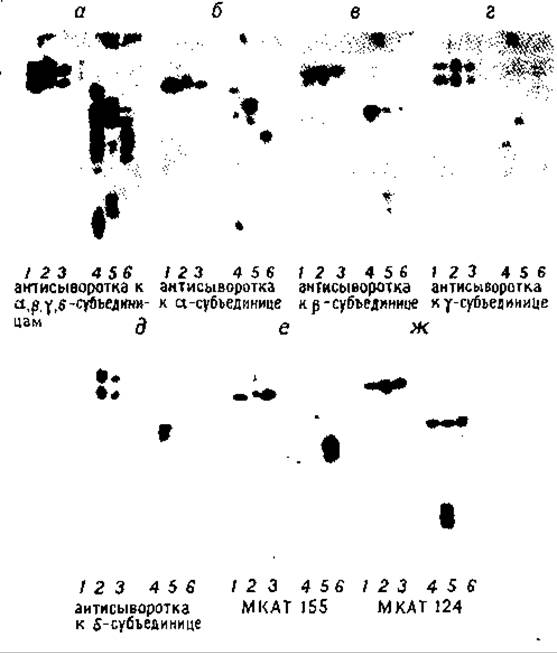
РИС. 7.2. Интактные субъединицы ацетилхолинового рецептора из Torpedo (I), Electrophorus (II), мышц быка (III) и их гидролизаты протеазой V8 разделены на 15%-ном ПААГ в присутствии ДСН и перенесены электрофоретически на диазотированную бумагу. 1 — I (10 нг); 2 — II (20 нг); 3 — III (50 нг); 4 — гидролизат I (40 нг, 1 : 10 по массе); 5 — гидролизат II (80 нг, 1 : 10); 6 — гидролизат III (100 нг, 1:10). Обнаружение с помощью смеси равных количеств антисывороток к а,ß,у,δ-субъединицам I (а), антисывороток к а,ß,у,δ-субъединицам соответственно (б—д) и моноклональных антител (МКАТ) к а- и ß-субъединицам (е и ж) [8]. (С разрешения Chemical Society.)
Образцы белков, предназначенные для пептидного картирования, часто находятся в больших объемах. Для их концентрирования и одновременного удаления нежелательных компонентов буферов белок осаждают обработкой 50%-ной (масс./об.) трихлороуксусной кислотой (для осаждения большинства белков достаточно 2 ч при 4 °С) и отделяют центрифугированием. При работе с очень небольшими количествами радиоактивно меченных белков для «затравки» и полноты осаждения добавляют немеченный («холодный») белок-носитель (например, иммуноглобулин). Поскольку ни белок-носитель, ни протеаза не содержат метки, их содержание в растворе лимитируется только объемом нанесения образца на пластинку (0,25—5 мкл). Общее содержание белка >100 мкг в пробе нежелательно, поскольку приводит к образованию размытых полос на карте и достаточно большая доля радиоактивного материала может остаться на старте.
Образец белка растворяют или суспендируют в летучем буфере (0,5 мл 50 мМ бикарбоната аммония), к которому добавляют фермент (для радиоактивных образцов 1—50 мкг, для немеченых белков в соотношении 1 : 100). Смесь оставляют на 6— 24 ч при 37 °С. Иногда для полноты гидролиза бывает необходимо добавить вторую порцию трипсина, поскольку вследствие автопротеолиза этот фермент теряет активность во времени.
Радиоактивно меченные белки в кусочках геля уравновешивают аммонийбикарбонатным буфером и проводят протеолиз, как описано выше. Согласно некоторым методикам до ферментативного гидролиза, предлагается провести стадию алкилирования или окисления белка [26, 61]. Образующиеся небольшие фрагменты, более растворимые, чем исходный белок, пассивно элюируются с геля. Супернатантную жидкость концентрируют лиофилизацией, при этом удаляются летучие компоненты буфера.
Высушенную смесь коротких пептидов затем растворяют в небольшом объеме (10 мкл) электрофоретического буфера и наносят на стеклянные пластинки, покрытые слоем силикагеля или целлюлозы толщиной 0,1 — 1 мм. К сожалению, коммерчески доступные пластинки часто имеют неровное покрытие, что может быть обнаружено просматриванием пластинки на свет (темные полосы или пятна свидетельствуют о различной толщине слоя носителя). Забракованные пластинки можно использовать для предварительных проб: установления времени пробега, величины pH в электрофорезе или состава хроматографической системы растворителей. В качестве носителя могут быть использованы листы бумаги ватман 3 ММ [36, 37]. Несмотря на значительные потери пептидов в результате адсорбции, хроматография па бумаге применяется для контроля гомогенности пептидов (разд. 7.3.2.1).
Раствор образца следует наносить маленькими порциями (0,25 мкл) так, чтобы стартовая точка была как можно более компактной. Каждая порция раствора быстро высушивается с помощью фена или вентилятора. На пластинку наносят также несущие заряд окрашенные соединения, они мигрируют на определенное расстояние и служат для визуальной оценки процесса. Для электрофореза применяют буферы, содержащие пиридин, уксусную кислоту, воду, иногда бутанол [26, 60] (табл. 7.1). Для получения различных разделяющих систем можно варьировать pH раствора. Удобно операции осуществлять в следующей последовательности: сначала электрофорез, а потом хроматографию; однако такой порядок не обязателен.
Таблица 7.1. Условия двумерного разделения триптических пептидов на пластинках с тонким слоем силикагеля
|
Пластинки 200X200 мм с толщиной слоя 0,1—0,25 мм. |
|
Первое направление: электрофорез |
|
pH 3,5 пиридин — уксусная кислота — вода (2:20:978), 1000 В, 45 мин |
|
pH 6,5, пиридин — уксусная кислота — вода (100:3:897), 1000 В, 40 мин |
|
pH 4,7, бутанол — пиридин — уксусная кислота — вода (2:1:1: 18) |
|
Второе направление: хроматография при 25 °С |
|
Хлороформ — метанол — аммиак (2:2:1) |
|
Пропанол — аммиак (7:3) |
|
Бутанол — пиридин — уксусная кислота — вода (97 : 75 : 15 : 60), pH 5,3 |
Электрофоретическая пластинка осторожно увлажняется соответствующим буфером так, чтобы не смазались точки нанесения образца, и включается напряжение 1000 В на 40—90 мин (для пластинки 200X200 мм). Следует указать, что этот эксперимент небезопасен и все электрические приборы время от времени должны проверять квалифицированные специалисты. По окончании электрофореза пластинку снимают и высушивают в течение ночи в вытяжном шкафу. Поскольку используемые растворители летучи, при полном высушивании не должно оставаться запаха уксусной кислоты. При сравнительном анализе нескольких образцов очень важно, чтобы электрофоретическая и хроматографическая процедуры повторялись в абсолютно одинаковых условиях.
Характер поведения пептидов в хроматографическом процессе определяется составом применяемой системы растворителей. Повышение содержания в ней органических компонентов увеличивает относительную подвижность гидрофобных пептидов, поскольку стационарная фаза гидрофильна. Широкое применение нашли несколько систем растворителей [26, 60], по для оптимизации условий разделения в конкретных случаях используются разнообразные варианты (табл. 7.1).
Для хроматографирования во втором направлении пластинки помещают в стеклянные камеры. Точки нанесения образцов должны быть на —1 см выше уровня растворителя. Процесс продолжают до тех пор, пока растворитель достигнет или почти достигнет верхнего края пластинки (для пластинки размером 200X200 мм на это требуется около 5 ч). После высушивания на воздухе, если анализировались меченые образцы, проводят авторадиографию прямым путем или в случае изотопов с низкой энергией опрыскивают хроматограмму раствором сцинтиллятора с последующей флуорографией [3].
Детекция немеченых пептидов осуществляется различными способами [55], но чаще всего с помощью флуорескамина в ацетоне [60]; образующиеся флуоресцирующие пятна далее исследуют под УФ-светом (366 нм) (гл. 8). Может проводиться непрерывная регистрация результатов в виде авторадиографических оттисков на рентгеновской пленке или фотографирования пластинок после обработки флуоресцентным реагентом (в течение <24 ч).
Если в результате сравнения полученных карт возникают сомнения относительно гомологичности двух белков, процедуру повторяют, нанося на одну пластинку равное количество каждого образца. После проявления хроматограммы совпадения или различия в положении пятен становятся более очевидными.
Проблемы, которые возникают при двумерном картировании, связаны с низким качеством пластинок (неравномерное покрытие), неправильным нанесением образца (избыточное количество белка или слишком малая его радиоактивность) и неоптимальными условиями проведения гидролиза белка и собственно процессов хроматографии и электрофореза. Следует иметь в виду, что хотя большинство пептидов в электрофоретическом буфере заряжены положительно, в смеси могут находиться и пептиды с отрицательным зарядом. Поэтому рекомендуется выполнить дополнительный эксперимент, для чего образец наносят по центру в нижней части пластинки и проводят электрофорез в течение 30 мин. Затем описанными приемами определяют подвижность пептидов в первом направлении и оптимальную продолжительность процесса в препаративном опыте. На рис. 7.3 приведено несколько двумерных пептидных карт для демонстрации результатов, которые могут быть достигнуты этим методом.
7.3.2.1. Контроль степени чистоты пептидов. Гомогенность пептидов может быть определена двумерным разделением на большом листе бумаги ватман 3 ММ (460X570 мм). Б первом направлении — электрофорез в системе: пиридин — уксусная кислота — вода (10:100:2800), pH 3,6, 2,2 кВ, 80 мин (могут быть использованы системы с другими pH). Во втором направлении — хроматография в системе w-бутанол — пиридин уксусная кислота — вода (150:100:30:120). Локализация пятен флуорескамином или реагентом нингидрин-кадмий.