Основы биохимии - Филиппович Ю. Б. 1999
Ферменты
Классификация ферментов и характеристика некоторых групп
По первой в истории изучения ферментов классификации их делили на две группы: гидролазы, ускоряющие гидролитические реакции, и десмолази, ускоряющие реакции негидролитического распада. Затем была сделана попытка разбить ферменты на классы по числу субстратов, участвующих в реакции. В соответствии с этим ферменты классифицировали на три группы. 1. Катализирующие превращения двух субстратов одновременно в обоих направлениях: А + В ⇄ С + D. 2. Ускоряющие превращения двух субстратов в прямой реакции и одного в обратной: А + В ⇄ С. 3. Обеспечивающие каталитическое видоизменение одного субстрата как в прямой, так и в обратной реакции: А ⇄ В.
Одновременно развивалось направление, где в основу классификации ферментов был положен тип реакции, подвергающейся каталитическому воздействию. Наряду с ферментами, ускоряющими реакции гидролиза (гидролазы), были изучены ферменты, участвующие в реакциях переноса атомов и атомных групп (феразы), в изомеризации (изомеразы), расщеплении (лиазы), различных синтезах (синтетазы) и т. д. Это направление в классификации ферментов оказалось наиболее плодотворным, так как объединяло ферменты в группы не по надуманным, формальным признакам, а по типу важнейших биохимических процессов, лежащих в основе жизнедеятельности любого организма. По этому принципу все ферменты делят на 6 классов.
1. Оксидоредуктазы — ускоряют реакции окисления — восстановления. 2. Трансферази — ускоряют реакции переноса функциональных групп и молекулярных остатков. 3. Гидролазы — ускоряют реакции гидролитического распада. 4. Лиазы — ускоряют негидролитическое отщепление от субстратов определенных групп атомов с образованием двойной связи (или присоединяют группы атомов по двойной связи). 5. Изомеразы — ускоряют пространственные или структурные перестройки в пределах одной молекулы. 6. Лигазы — ускоряют реакции синтеза, сопряженные с распадом богатых энергией связей. Эти классы и положены в основу новой научной классификации ферментов.
1. Оксидоредуктазы. К классу оксидоредуктаз относят ферменты, катализирующие реакции окисления — восстановления. Общая схема их может быть представлена следующим образом:

Окисление протекает как процесс отнятия атомов Н (электронов) от субстрата, а восстановление — как присоединение атомов Н (электронов) к акцептору. Если обозначить акцептор буквой А, а субстрат — В, то уравнение реакции окисления — восстановления при участии оксидоредуктаз примет такой вид:
![]()
Характерной особенностью деятельности оксидоредуктаз в живой клетке является их способность образовывать системы (так называемые цепи окислительно-восстановительных ферментов), в которых осуществляется многоступенчатый перенос атомов водорода или электронов от первичного субстрата к конечному акцептору, которым является, как правило, кислород, так что в результате образуется вода.
Те оксидоредуктазы, которые переносят атомы Н или электроны непосредственно на кислородные атомы, носят название аэробных дегидрогеназ или оксидаз. В отличие от них оксидоредуктазы, переносящие атомы Н и электроны от одного компонента окислительной цепи ферментрв к другому без передачи их на кислородные атомы называют анаэробными дегидрогеназами или редуктазами.
Если фермент катализирует реакцию отнятия Н непосредственно от окисляемого вещества (первичного субстрата), то его называют первичной дегидрогеназой. Если фермент ускоряет снятие водородных атомов со вторичного субстрата, который получил атомы Н при посредстве первичной дегидрогеназы (вторичным субстратом может быть кофермент самой первичной оксидоредуктазы), его называют вторичной дегидрогеназой (см. гл. X).
Другая особенность оксидоредуктаз состоит в том, что, будучи двухкомпонентными ферментами с весьма ограниченным набором активных групп (коферментов), они способны ускорять большое число самых разнообразных окислительно-восстановительных реакций. Это достигается за счет того, что один и тот же кофермент способен соединяться со многими апоферментами, образуя каждый раз оксидоредуктазу, специфичную по отношению к тому или иному субстрату или акцептору.
Еще одна, пожалуй, главная особенность оксидоредуктаз заключается в том, что они ускоряют протекание химических процессов, связанных с высвобождением энергии. Последняя используется как для обеспечения синтетических процессов в организме, так и для других нужд.
В природных объектах обнаружено около пятисот индивидуальных оксидоредуктаз. Наиболее распространены оксидоредуктазы, содержащие в качестве активной группы никотинамидадениндинуклеотид, или НАД+ (о строении нуклеотидов см. гл. VI):
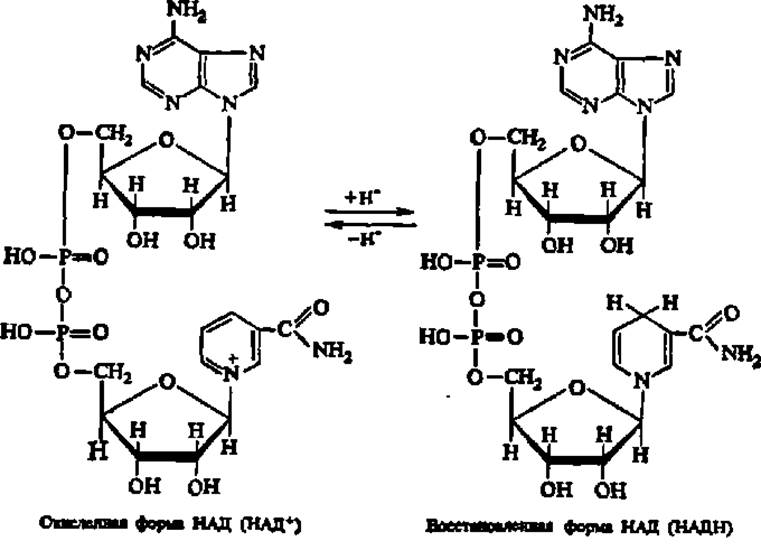
Более половины известных в настоящее время оксидоредуктаз содержат НАД+ в качестве кофермента. Соединяясь с тем или иным специфическим белком и образуя таким образом двухкомпонентный фермент, который сокращенно называют пиридинпротеином, НАД+ резко усиливает свою способность восстанавливаться по ядру никотинамида. В результате пиридинпротеины способны отнимать от субстратов (спирты, альдегиды, дикарбоновые и кетокислоты, амины и др.) атомы Н в виде гидрид-ионов (Н~) и протонов (Н+), окисляя, таким образом, указанные соединения. Все пиридинпротеины являются анаэробными дегидрогеназами, т.е. не передают снятые с субстрата атомы водорода на кислород, а посылают их на ближайший в окислительной цепи другой фермент.
Рассмотрим строение и механизм действия одного из пиридинпротеинов — алкогольдегидрогеназы из печени животных. Это белок с М = 73000, состоящий из двух субъединиц, каждая из которых несет молекулу НАД+ и атом Zn. В процессе отнятия атомов Н от спирта образуется тройной апофермент-кофермент-субстратный комплекс, удерживаемый Zn2+, Строение этого комплекса и механизм реакции окисления спирта в альдегид представлены на рис. 53. Непосредственно к никотинамидадениндинуклеотиду от молекулы спирта переходит один атом водорода в виде гидридного иона (H+), т.е. атома водорода, несущего дополнительный электрон. Второй атом водорода, отнимаемый от молекулы спирта, наоборот, теряет электрон, превращаясь в протон (Н+), и поступает в реакционную среду. Поэтому уравнение реакции окисления спирта при участии НАД+ записывают так:
![]()
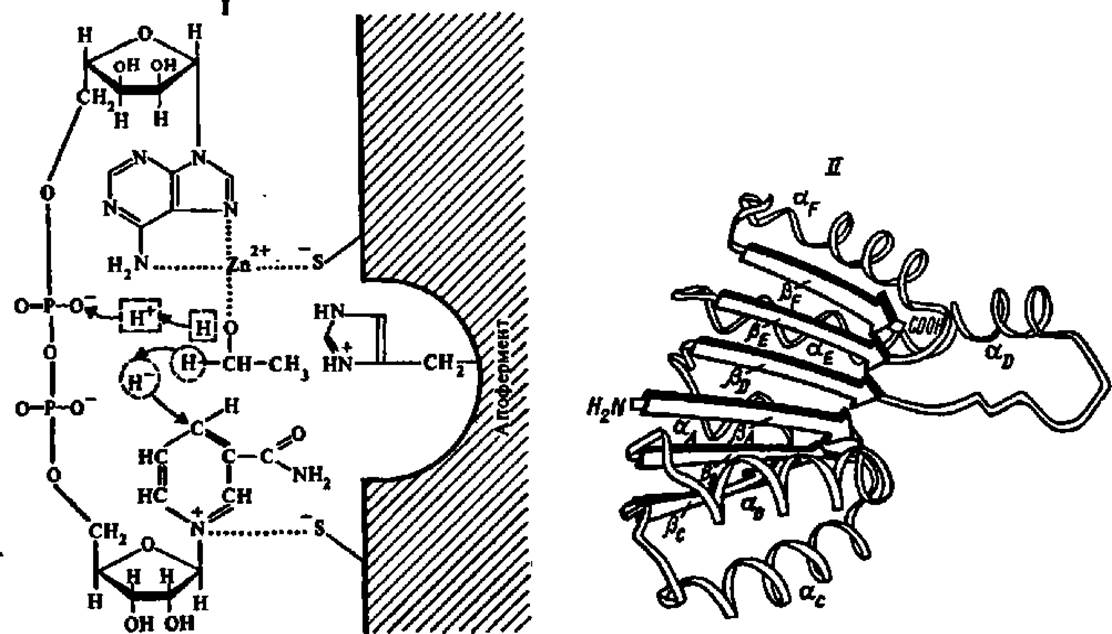
Рис. 53. Механизм действия алкогольдегидрогеназы
НАД* удерживается на поверхности белковой молекулы связями, возникающими между положительно заряженным атомом азота пиридинового цикла и отрицательно заряженным атомом серы (из HS-группы), а также между атомами азота пуринового цикла и атомом серы через посредство Zn2+. Молекула спирта присоединяется к активному центру фермента за счет координационной связи между атомом О и Zn2+. Каталитическую функцию в переносе атомов водорода от молекулы спирта к НАД+ выполняет имидазольный радикал гистидина. К пиридиновому ядру НАД+ присоединяется атом водорода, ранее находившийся в связи с атомом углерода, несущим спиртовую группу. Атом водорода спиртовой группы протонируется (І). Присоединение НАД+ к апоферменту происходит по нуклеотидсвязывающему домену (II), составленному из а-спиралей и ß-слоев. Структура нуклеотидсвязывающего домена близка у всех НАД-зависимых дегидрогеназ; к нижней части домена (ß-тяжи А, В и С) присоединяется фрагмент АМФ, а к верхней (ß-тяжи D, Е и F) — фрагмент никотинамидрибозофосфата молекулы НАД*
В любом случае НАД+ получает два электрона за счет присоединения гидридного иона (H).
Кроме НАД+ пиридинферменты содержат в качестве кофермента никотинамидадениндинуклеотидфосфат (НАДФ+). Этот кофермент является производным НАД+, у которого водород ОН-группы 2-го углеродного атома рибозы аденозина замещен на остаток фосфорной кислоты.
НАДФ+, соединяясь со специфическими белками, образует большую группу пиридинпротеинов, характеризующуюся своим набором субстратов. Механизм окисления при участии НАДФ+ в качестве кофермента аналогичен таковому при посредстве НАД+. Более того, НАДН и НАДФ+, равно как НАДФН и НАД+, при каталитическом участии специального фермента—трансгидрогеназы — способны обмениваться атомами водорода и электронами:
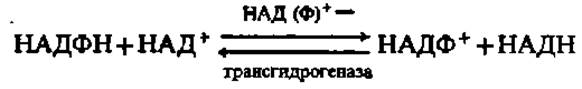
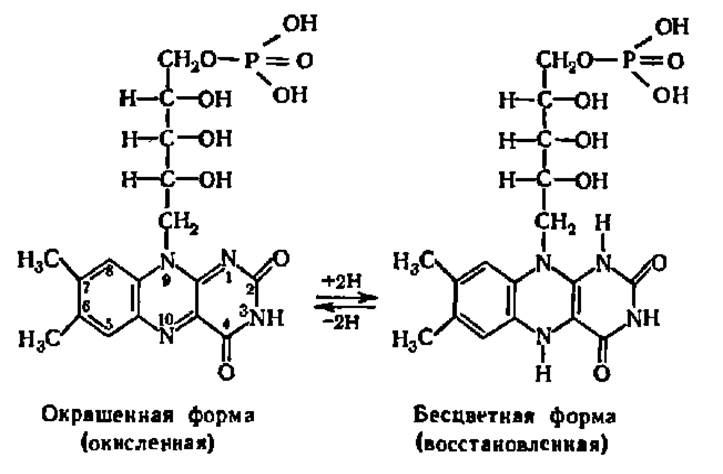
Партнером восстановленных форм пиридинпротеинов в оксидоредуктазной цепи, как правило, служат флавопротеины (ФП). Таким флавопротеином, например, является фермент, несущий в качестве активной группы фосфорилированный витамин В2. Окисленная форма этого флавопротеина (М = 52000) окрашена. Каждая молекула фермента несет молекулу рибофлавинфосфата (или флавинмононуклеотида, ФМН), способного принимать и отдавать два атома Н по атомам N изоаллоксазинового кольца:
Другим коферментом в флавопротеинах является флавинадениндинуклеотид (ФАД):
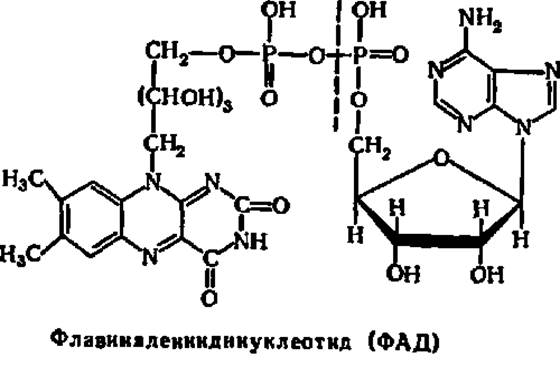
ФМН и ФАД, соединяясь с различными апоферментами, дают начало приблизительно тридцати флавопротеинам, отличающимся различной специфичностью по отношению к субстратам.
Основная функция флавопротеинов — перенос электронов (атомов Н) от восстановленных пиридинпротеинов к другим компонентам окислительновосстановительной цепи, т.е. ФП в большинстве случаев являются вторичными дегидрогеназами. Однако некоторые флавопротеины, особенно с ФАД в качестве кофермента, могут непосредственно снимать атом Н с субстрата.
Коферментами оксидоредуктаз являются также хиноны. Так, соединяясь с белком, убихиноны образуют убихинонпротеин, являющийся важной составной частью ансамблей оксидоредуктаз, при посредстве которых осуществляется перенос атомов Н и электронов.
Убихиноны являются производными бензохинона и обладают боковой цепью, составленной из большого числа изопреноидных остатков:
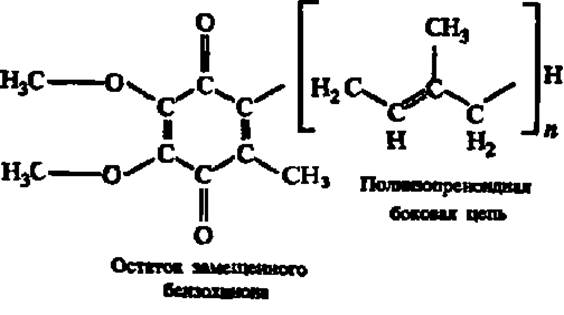
Число изопреноидных фрагментов в боковой цепи (и) колеблется от б до 10. Установлено, что убихиноны принимают участие в окислительно-восстановительных процессах в организме, осуществляя передачу атомов Н:
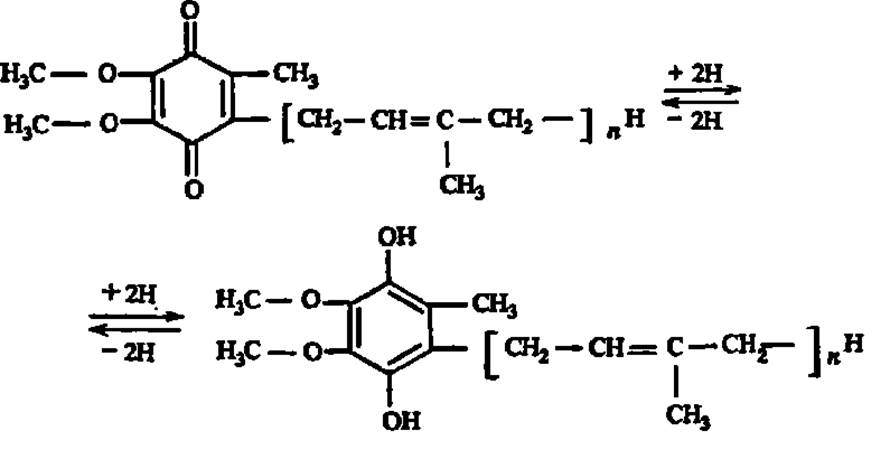
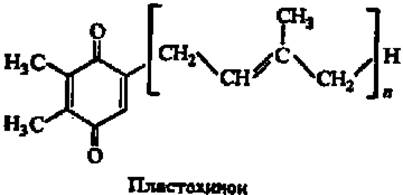
В растениях эту функцию выполняет похожее на убихинон соединение — пластохинон:
Наиболее сложный, но и самый распространенный вариант окислительновосстановительного процесса в клетке состоит в окислении атомов Н, снятых с субстрата, при посредстве цитохромной системы.
Цитохромную систему образуют несколько оксидоредуктаз, имеющих в качестве простетических групп железопорфирины (рис. 54). Еще в 1915 г., двадцатью годами ранее О. Варбурга, на возможную роль железосодержащих белков в биологическом окислении обратили внимание А. Я. Данилевский и Б. П. Соловцов. Соединяясь с белками различного строения, железопорфирины 4 типов (А, В, С и D) дают начало семейству хромопротеинов, объединяемых под общим названием — цитохромы. Сейчас известно несколько десятков цитохромов, и список их непрерывно пополняется. Каждый индивидуальный цитохром обозначают строчной латинской буквой а, b, с и d с соответствующим порядковым индексом, например b1, b2, b3 и т.д., а класс цитохрома — прописной латинской буквой А, В, С или D. Принадлежность цитохрома к определенному классу определяется строением простетической группы (железопорфирина), а окончательная индивидуальность — строением апофермента (белка). В последнее время предпочитают наряду с порядковым номером цитохрома указывать характерную длину волны, при которой отмечается поглощение в видимой части спектра (например, цитохром b6 или b563, найденный в хлоропластах, и т. п.).
Первичная структура ряда цитохромов выяснена: оказалось, что их видовая специфичность связана с небольшими различиями в чередовании аминокислот. Эти данные принципиально важны для понимания природы видовой и иной специфичности ферментов: видимо, она определяется различиями прежде всего в первичной структуре апоферментов. Цитохромы b1, b2, b3 и т. д., содержащие одну и ту же простетическую группу, отличаются Друг от друга именно по этому признаку.
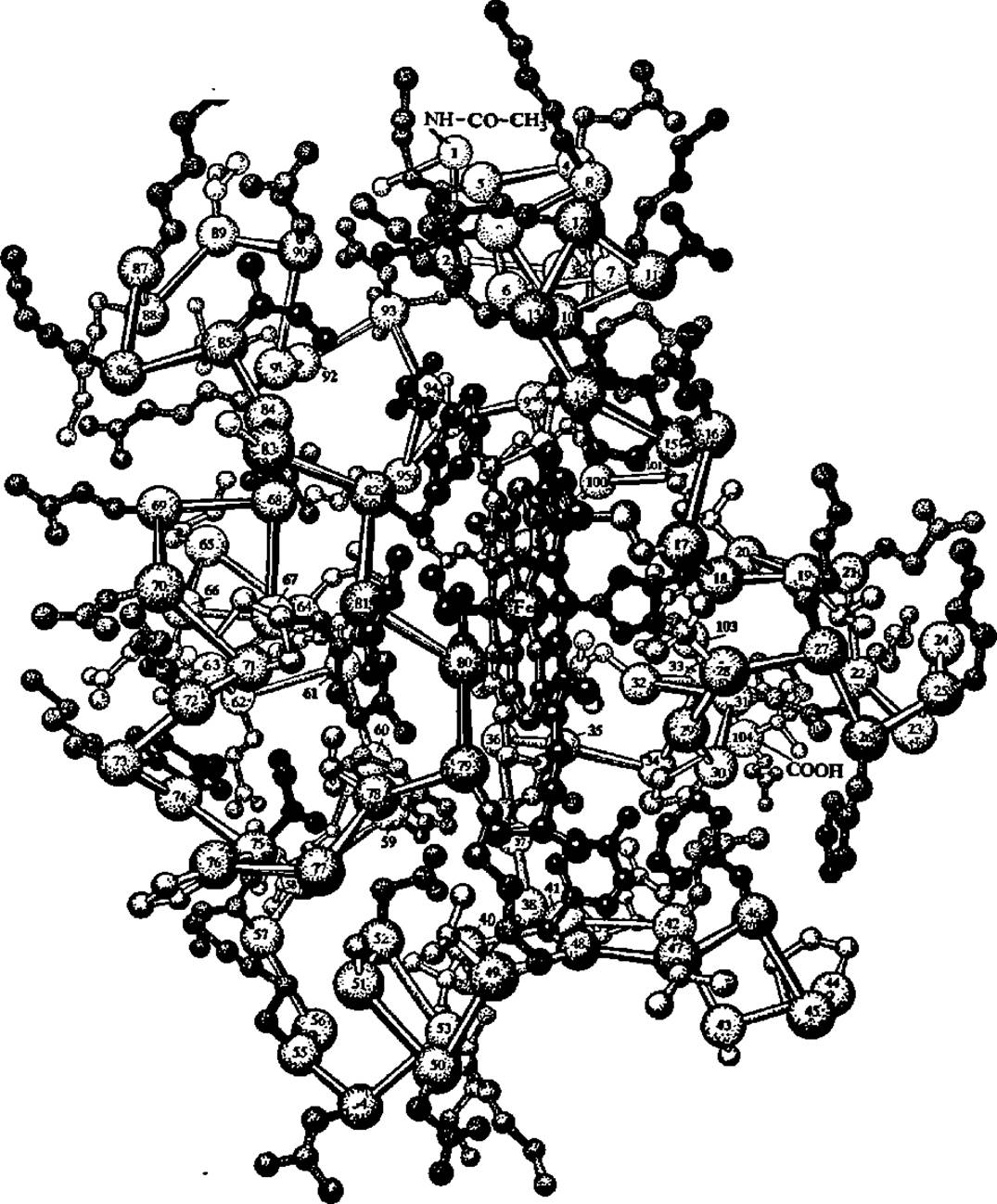
Рис. 54. Строение цитохрома с из сердечной мышцы лошади
Простетическая группа цитохрома с представлена железопорфирином (с атомом Fe2+ в центре). Связь его с белком осуществляется за счет взаимодействия винильных радикалов гема с HS-группами 14-го и 17-го остатков цистеина полипептидной цепи. Аминогруппа N-концевого глицина ацетилирована. На объемной модели цитохрома с цифрами указаны номера аминокислотных остатков, каждый из которых также представлен пространственной структурой
На рис. 54 приведена структура цитохрома с из сердца лошади. Молекулярная масса цитохрома с невелика — порядка 13 000. При увеличении в 1 млн. 300 тыс. раз (электронная микроскопия) видно, что полипептидная цепь цитохрома с свернута в ос-спираль длиной в 12—15 нм, которая, в свою очередь, закручена в спираль второго порядка с диаметром в 4—5 нм, так что группа гема оказывается изолированной внутри обвивающей ее полипептидной цепи. Кольцеообразные молекулы легко образуют более крупные агрегаты. Строение остальных цитохромов изучено менее детально. Однако известно, что молекулярные массы некоторых из них более высоки. Способность агрегировать друг с другом, по-видимому, одно из свойств, присущих молекулам цитохромов.
Именно поэтому цитохромы образуют цитохромную систему, представляющую собой упорядоченное сочетание в едином комплексе различных цитохромов, например b, с и а.
Цитохромная система способна принимать электроны, снятые с атомов Н восстановленного убихинона (УХ). Она передает электроны далее по цепи цитохромов и, наконец, на кислородный атом; последний, соединяясь с ионизированными атомами Н, образует молекулу Н2O.
В цепи цитохромов каждый из индивидуальных цитохромов занимает строго определенное место. Простейший вариант цитохромной системы приведен на следующей схеме:
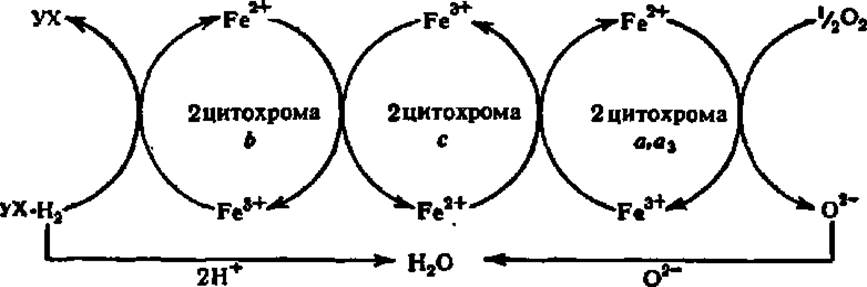
Как видно из схемы, передача электронов в цитохромной цепи осуществляется за счет изменения валентности атома Fe порфиринового ядра. Из всех цитохромов только цитохром с, а3 передает электроны на кислород. Поэтому именно он завершает цепь цитохромов и носит название цитохромоксвдазы. Кроме атомов Fe (в составе гема) цитохром а, а3 содержит также атомы Сu, с которыми связывают его окислительные свойства (см. гл. X, рис. 132).
Таковы характерные черты действия некоторых важнейших оксидоредуктаз и окислительно-восстановительных систем, обеспечивающих превращение ряда веществ в клетке.
2. Трансферази. В этот класс входят ферменты, ускоряющие реакции переноса функциональных групп и молекулярных остатков от одного соединения к другому. Это один из наиболее обширных классов: он насчитывает около 500 индивидуальных ферментов. В зависимости от характера переносимых группировок различают фосфотрансферазы, аминотрансферазы, гликозилтрансферазы, ацилтрансферазы, трансферазы, переносящие одноуглеродные остатки (метилтрансферазы, формилтрансферазы), и др.
Фосфотрансферазы. Сюда относятся ферменты, ускоряющие реакцию переноса остатка фосфорной кислоты. Эта реакция имеет исключительно важное значение для жизнедеятельности организма, обеспечивая превращение ряда органических соединений в фосфорные эфиры, обладающие повышенной химической активностью и более легко вступающие в последующие реакции. Перенос фосфатных групп идет на спиртовые, карбоксильные, азотсодержащие, фосфорсодержащие и другие группы тех или иных органических соединений. В соответствии с этим среди фосфортрансфераз различают несколько подподклассов.
Донором фосфатных остатков является в большинстве случаев аденозинтрифосфорная кислота (АТФ), но возможны и другие их источники. К фосфотрансферазам относится, например, гексокиназа — фермент, ускоряющий перенос остатка фосфорной кислоты от молекулы АТФ к глюкозе (с этой реакции обычно начинается преобразование глюкозы):
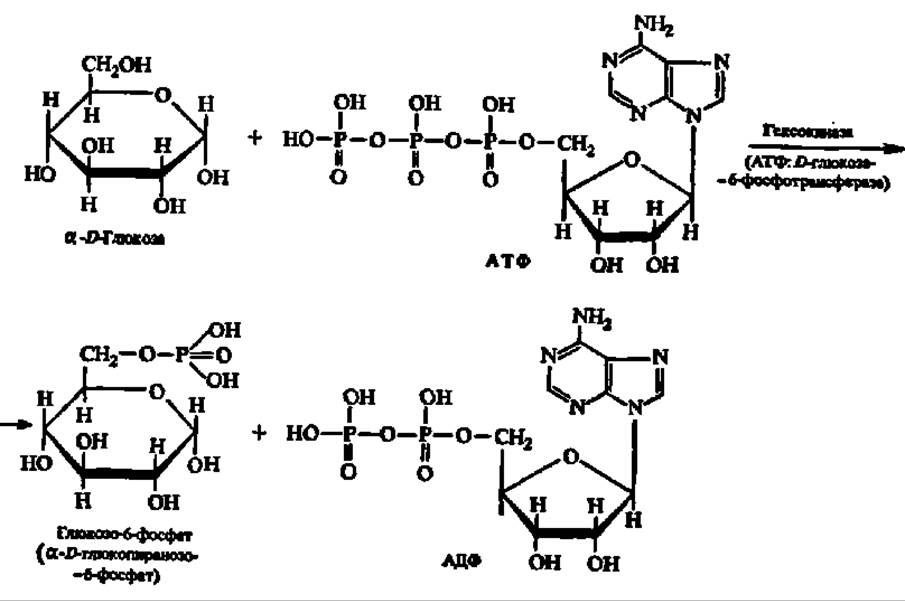
Гексокиназа распространена повсеместно. Особенно хорошо изучена гексокиназа дрожжей. Ее молекула (М = 96 000) составлена из 4 субъединиц. Мультимер устойчив при pH 5. При снижении или повышении pH раствора молекула гексокиназы распадается на 4 протомера (М = 24 000), лишенных фосфо- трансферазной активности. В соответствии с двойственной природой субъединиц в мультимерах обнаружено 5 изозимов гексокиназы.
Преобразование многих других моносахаридов тоже начинается с их фосфорилирования при посредстве фосфотрансфераз: так обстоит дело в случае ß- D-фруктозы, ß-D-рибозы (см. с. 338) и ряда других сахаров.
Особое внимание в последнее время уделяют изучению фосфотрансфераз, обеспечивающих перенос остатка фосфата с АТФ на белки, — протеинкиназам. Они переносят фосфат на радикалы сер, тре, тир, лиз и гис ряда белков, в результате чего резко изменяется биологическая активность последних. Это, в свою очередь, сказывается на интенсивности протекания химических процессов в организме, т. е. на регуляции обмена веществ (см. гл. XIII).
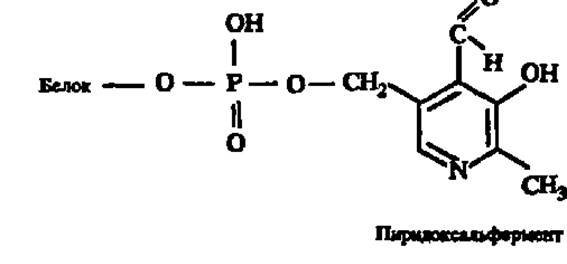
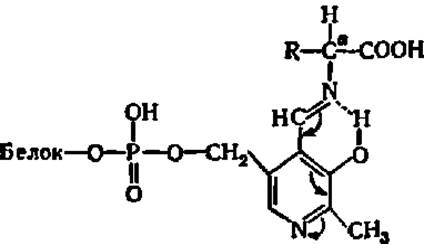
Аминотрансферазы. Эти ферменты ускоряют реакцию переаминирования аминокислот с кетокислотами и очень важны для обеспечения биосинтеза аминокислот. Аминотрансферазы двухкомпонентны: простетической группой их во всех случаях является пиридоксальфосфат, ковалентно присоединенный к апоферменту через свою альдегидную группу (см. рис. 55, Б) и ионной связью — через остаток фосфорной кислоты:
Механизм реакции переаминирования сейчас хорошо выяснен, а сама реакция открыта еще в 1937 г. А. Е. Браунштейном и М. Г. Крицман.
Для удобства пиридоксальфермент обозначим при помощи следующей структуры: ![]() сохранив только функционально значимую альдегидную группу его кофермента.
сохранив только функционально значимую альдегидную группу его кофермента.
На. первой стадии ферментативного катализа простетическая группа фермента (для простоты принято, что она свободна, а не соединена альдиминной связью с апоферментом через Б-аминогруппу радикала лиз) взаимодействует с аминокислотой, подвергающейся переаминированию. Реакция идет по аминогруппе аминокислоты и альдегидной группе остатка пиридоксальфосфата:
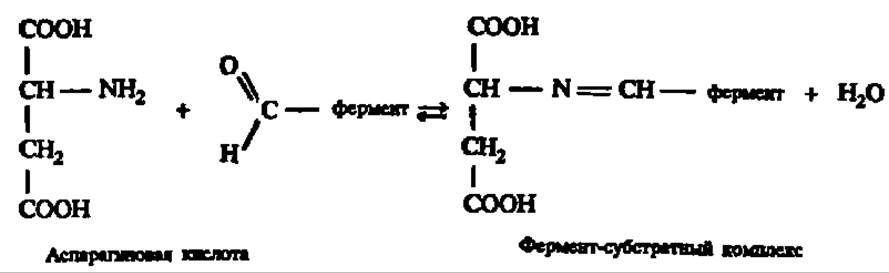
На второй ступени катализа идет преобразование субстрата, выражающееся в данном случае в таутомерной перегруппировке:
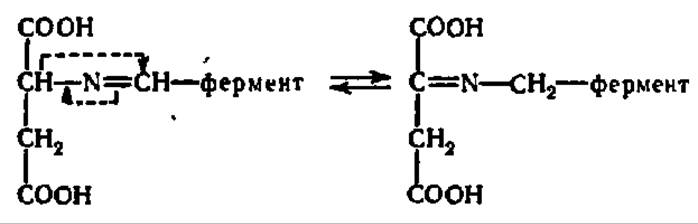
Эта перегруппировка осуществляется при участии имидазолсодержащих радикалов остатков гис, входящих в состав каталитического центра фермента:
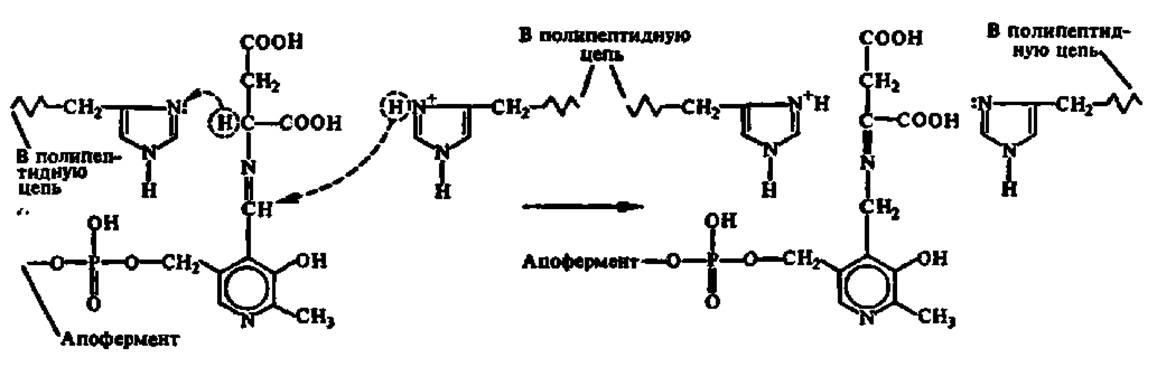
В результате последующего гидролиза освобождаются кетокислота и фермент в виде пиридоксаминофермента:
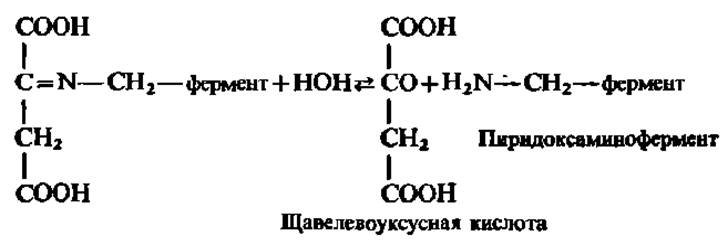
Далее между пиридоксаминоферментом и другой кетокислотой вновь возникает фермент-субстратный комплекс:
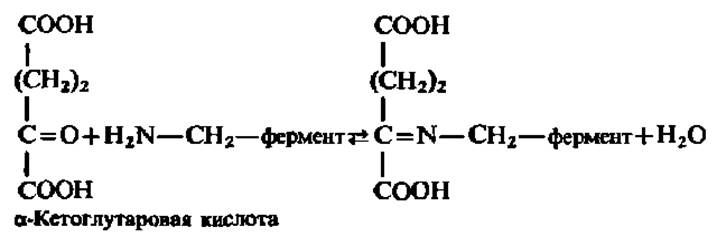
Субстрат в нем снова подвергается преобразованию за счет таутомерного превращения:
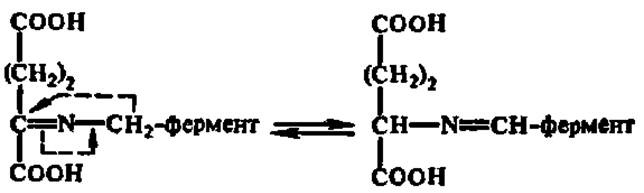
Полученное соединение гидролизируется, и возникает новая аминокислота:
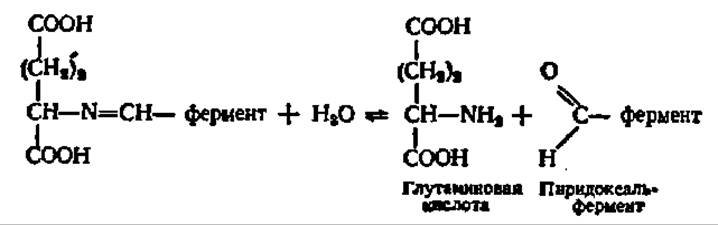
Следовательно, в результате серии реакций, включающих в себя попеременное образование фермент-субстратных комплексов, аспарагиновая кислота переходит в щавелевоуксусную, а а-кетоглутаровая — в глутаминовую. Это выражается следующим суммарным уравнением:
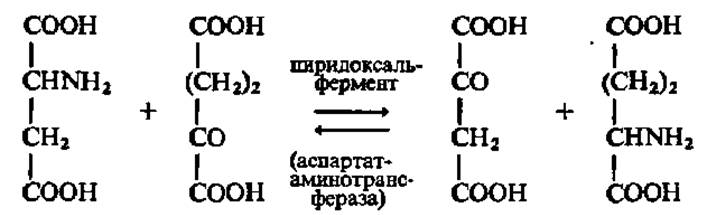
Центральную роль в пиридоксалевом катализе играет смещение электронной плотности в фермент-субстратном комплексе:
В результате у а-углеродного атома аминокислотного остатка ослабляются связи с заместителями (азотом, СООН-группой и др.), вследствие чего легко осуществляется разрыв соответствующих связей.
Аспартатаминотрансфераза имеет молекулярную массу, равную 93000, и состоит из двух идентичных субъединиц (М = 46 500), каждая из которых соединена с молекулой пиридоксальфосфата. При разбавлении растворов аспартатаминотрансферазы ее димеры распадаются на каталитически активные мономеры. Благодаря исследованиям главным образом советских ученых (А. Е. Браунштейна с сотр., Ю. А. Овчинникова с сотр. и Б. К. Вайнштейна с сотр.) выяснены первичная и третичная структуры этого фермента, строение и детальный механизм функционирования его активного центра. Субъединица цитозольного изофермента аспартатаминотрансферазы из сердца свиньи (другой изофермент локализован в митихондриях) представлена полипептидной цепью из 412 аминокислотных остатков. Значительная часть ее находится в а-спиральной конформации, а в обособленном участке глобулы расположен коферментсвязывающий домен, где локализован активный центр (рис. 55). Характерно, что коферментсвязывающий домен пиридоксальферментов очень похож на нуклеотидсвязывающий домен НАД+- и НАДФ+-зависимых дегидрогеназ (см. рис. 53, II), что объясняется, видимо, присутствием пиридинового цикла в составе того и другого кофермента.
Поскольку аспартатаминотрансфераза состоит из двух субъединиц и несет, следовательно, два остатка пиридоксальфосфата, в реакции переаминирования субъединицы работают согласованно, со сдвигом по фазе в использовании энергии, необходимой для осуществления химических преобразований; вследствие этого димерная структура фермента дает существенный выигрыш в осуществлении каталитического процесса.
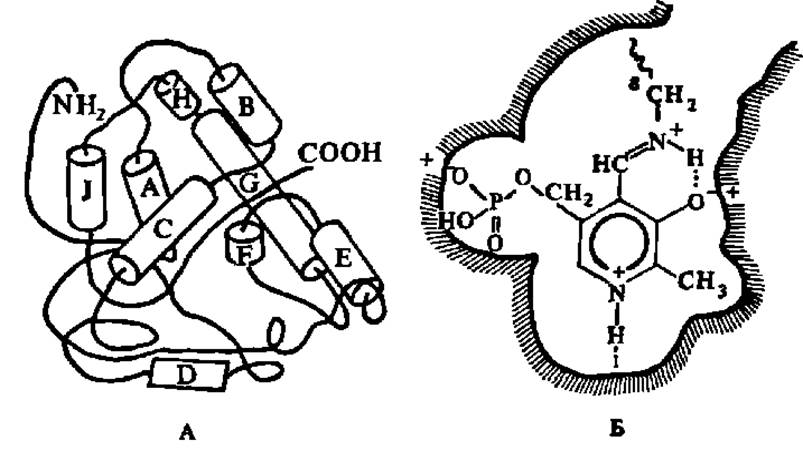
Рис. 55. Один из возможных вариантов третичной структуры аспартатаминотрансферазы (А) и строение ее активного центра (Б)
На рис. Б видно, что пиридоксальфосфат соединен альдиминной связью с Е-аминогруппой остатка лизина в апоферменте; именно по этой альдиминной связи присоединяется аминогруппа аминокислоты, вытесняя оттуда ε-аминогруппу лизина
Гликозилтрансферазы. Эти ферменты ускоряют реакции переноса гликозильных остатков из молекул фосфорных эфиров или других соединений к молекулам моносахаридов, полисахаридов или иных веществ, обеспечивая главным образом реакции синтеза и распада олиго- и полисахаридов в животном и растительном мире. Ниже приведено уравнение реакции распада сахарозы при участии сахароза: ортофосфат-а-глюкозилтрансферазы, или сахарозофосфорилазы:
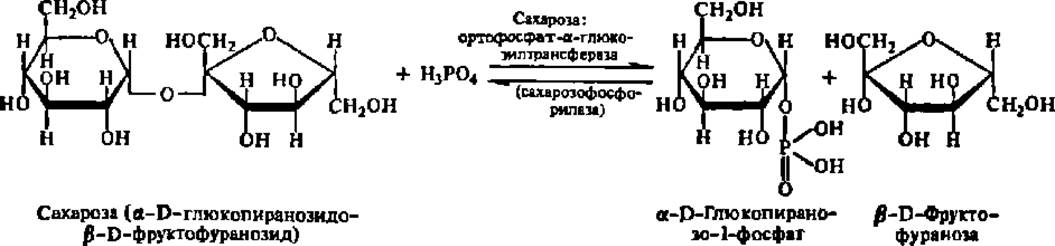
Аналогично этому действуют крахмалфосфорилаза, гликогенфосфорилаза и другие гликозилтрансферазы. В случае переноса гликозильных остатков на Н3РО4 этот процесс называют фосфоролизом, так как он формально аналогичен гидролизу, но вместо элементов воды по месту разрыва кислородного мостика присоединяются водород и фосфатная группа фосфорной кислоты (подробнее о гликогенфосфорилазе и механизме ее действия см. гл. VIII).
В последнее время выяснено, что перенос гликозильных остатков особенно легко осуществляется ферментами данной группы в тех случаях, когда субстратом служит нуклеозиддифосфатмоносахарид. Эта реакция представляет, видимо, основной путь природного синтеза олиго- и полисахаридов и будет детально рассмотрена в гл. VIII. Нуклеозиддифосфатсахара являются коферментами гликозилтрансфераз.
Ацилтрансферазы. Эти ферменты ускоряют перенос ацилов (остатков карбоновых кислот) на аминокислоты, амины, спирты и другие соединения. Универсальным источником ацильных групп во всех этих реакциях является ацил-коэнзим А, который с полным основанием можно рассматривать как активную группу ацилтрансфераз.
Чаще всего переносу в биологических объектах подвергается ацил уксусной кислоты — ацетил ![]()
Коэнзим А (см. формулу на с. 163), соединяясь с ацетильным остатком, который занимает место водорода в его HS-группе, образует ацетил-коэнзим А. Последний служит кофактором в соответствующей реакции переноса. Одним из примеров реакции трансацилирования является синтез ацетилхолина:
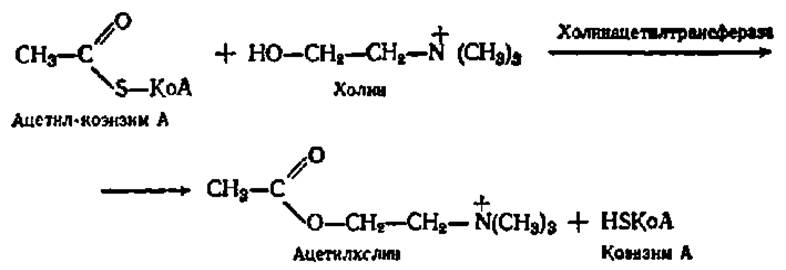
Важное значение среди трансфераз имеют ферменты, ускоряющие перенос одноуглеродных фрагментов (метальных, оксиметильных, формильных и т. п.), а также нуклеотидилтрансферазы, катализирующие перенос нуклеотидных остатков в процессе синтеза нуклеиновых кислот. Механизм их действия будет описан ниже.
3. Гидролазы. К классу гидролаз относят ферменты, ускоряющие реакции расщепления (а иногда и синтеза) органических соединений при участии воды: R'R"+HOH ⇄ R'H+R"OH. В зависимости от характера субстрата, подвергающегося гидролизу, гидролазы делят на ряд подклассов, среди которых наиболее важны следующие:
1) эстеразы, ускоряющие реакции гидролиза сложных эфиров;
2) гликозидазы, ускоряющие реакции гидролиза гликозидов, в том числе углеводов;
3) пептид-гидролазы, ускоряющие реакции гидролиза (а в особых случаях и синтеза) белков, пептидов и других соединений, содержащих пептидные связи;
4) гидролазы, действующие на С—N-связи, отличающиеся от пептидных (например, амидазы и т. п.). Всего в составе гидролаз насчитывают почти 500 ферментов.
Эстеразы. Эти ферменты катализируют реакции гидролиза сложных эфиров спиртов с органическими и неорганическими кислотами. Важнейшими подподклассами эстераз являются гцдролазы эфиров карбоновых кислот и фосфатазы. В качестве представителя первого подподкласса рассмотрим липазу.
Липаза ускоряет гидролиз внешних, т. е. а-сложноэфирных, связей в молекулах триацилглицеринов (жиров):
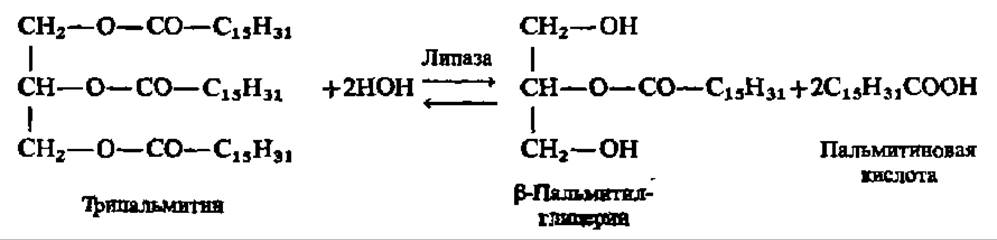
Механизм действия ряда эстераз детально изучен. Один из примеров рассмотрен в этой главе (см. раздел о механизме действия ферментов). Характеристика липаз дана в гл. IX.
Фосфатазы катализируют гидролиз фосфорных эфиров. Особенно широко распространены фосфатазы, действующие на сложные эфиры фосфорной кислоты и углеводов, например глюкозо-1-фосфатаза:
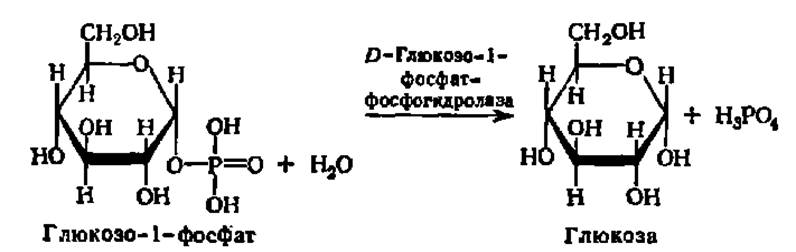
Действие фосфатаз проявляется в широком спектре pH от 3 до 9. Большинство из них обладает широкой субстратной специфичностью. Особенно важны для регуляции процессов жизнедеятельности протеинфосфатазы, обеспечивающие отщепление фосфата от фосфорилированных белков, вследствие чего изменяется их биологическая, в частности ферментативная, активность.
Гликозидазы. Эти ферменты ускоряют реакцию гидролиза гликозидов. В зависимости от того, на какой пространственный изомер (а или ß) действует фермент, его относят к а- или ß-гликозидазам. Таким образом, гликозидазы обладают ярко выраженной пространственной специфичностью. Кроме гликозидов, содержащих в качестве агликонов остатки одноатомных спиртов, субстратами, на которые распространяется действие тех или иных гликозидаз, являются олиго- и полисахариды. Из действующих на олигосахариды гликозидаз упомянем мальтазу (а-гликозидаза) и сахаразу (ß-гликозидаза). Они ускоряют соответственно гидролиз мальтозы и сахарозы:
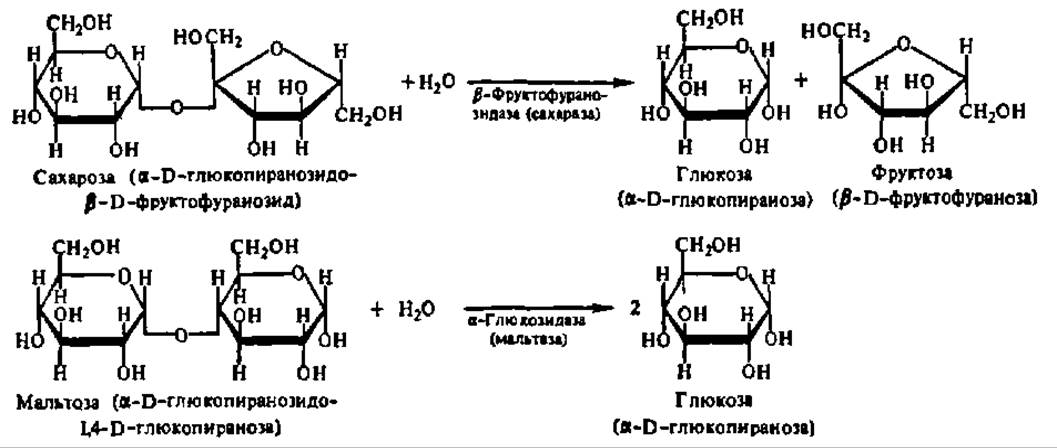
Из гликозидаз, действующих на полисахариды, наиболее известны амилазы. В природе существует несколько видов амилаз, ускоряющих реакции гидролиза гликозидных связей в молекуле крахмала с образованием глюкозы, мальтозы или олигосахаридов. Их характеристика и механизм действия рассмотрены в гл. VIII.
Гидролиз других природных полигликозидов: целлюлозы, инулина, ксилана и т. п. — также ускоряется соответствующими гликозидазами. Некоторые гликозидазы катализируют также реакции переноса гликозильных остатков, т. е. являются трансгликозидазами.
Пептид-гидролазы. Ферменты этого подкласса ускоряют гидролиз пептидных связей в белках и пептидах, а при определенных условиях также и образование пептидных связей, хотя этот путь синтеза белка не является физиологическим. Химизм процесса гидролиза белков и пептидов при участии пептидгидролаз можно выразить следующей схемой:
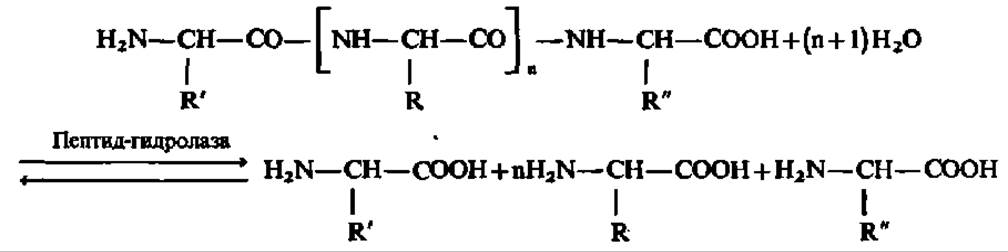
Среди пептид-гидролаз различают протеиназы или пептидил-пептидогидролазы, катализирующие гидролиз небольшого числа внутренних пептидных связей в белковой молекуле, в результате чего последняя распадается до пептидов. Они являются, следовательно, эндопептидазами. В отличие от этого пептид-гидролазы, называемые пептидазами, обеспечивают отщепление от пептидной цепи свободных аминокислот, будучи экзопептидазами.
Протеиназы в зависимости от механизма их действия на внутренние пептидные связи в белковой молекуле делят на 4 подподкласса: 1) сериновые протеиназы, несущие в активном центре радикалы сер и гис, обеспечивающие осуществление каталитического акта; представителями их являются химотрипсин и трипсин, выделяемые поджелудочной железой, субтилизин, продуцируемый бактериями, и др.; 2) тиоловые (цистеиновые) протеиназы, имеющие в активном центре остаток цис; к их числу принадлежат папаин из латекса дынного дерева Carica papaya, фицин из латекса фикуса, бромелаин из сока ствола ананаса, катепсин В — внутриклеточный фермент позвоночных и др.; 3) кислые (карбоксильные) протеиназы, имеющие оптимум pH ниже 5 и содержащие радикалы дикарбоновых аминокислот в активном центре; сюда относятся пепсин, выделяемый слизистой желудка, катепсин D, характеризующийся внутриклеточной локализацией, и ряд кислых протеиназ, продуцируемых разнообразными микроорганизмами; 4) металлопротеиназы, каталитическое действие которых зависит от присутствия ионов металлов (Са2+, Zn2+) в активном центре; примерами их могут служить коллагеназа и ряд протеиназ микробного происхождения (термолизин, компонент проназы и др.).
Пепсин, трипсин и химотрипсин выделяются железистыми клетками в виде неактивных проферментов — зимогенов: пепсиногена, трипсиногена и химотрипсиногена, так как их активные центры блокированы фрагментами полипептидной цепи, после гидролитического отщепления которых фермент приобретает активность. Это явление впервые было открыто в лаборатории И. П. Павлова.
Очень важной особенностью протеиназ является выборочный (селективный) характер их действия на пептидные связи в белковой молекуле. Так, пепсин избирательно ускоряет гидролиз пептидных связей, образованных фен и лей; трипсин — арг и лиз; химотрипсин — ароматическими аминокислотами; папаин — арг, лиз и фен и т. д. В результате индивидуальный белок под действием определенной пептидил-пептидогидролазы расщепляется всегда на строго ограниченное число пептидов. Это находит практическое использование при определении первичной структуры белков и имеет огромное значение для регуляции обмена веществ, так как многие продукты селективного гидролиза белков обладают высочайшей биологической активностью: именно этим путем из проферментов возникают ферменты, из предшественников гормонов — гормоны и рилизинг-факторы и т. п. Причина избирательного действия пептидпептидогидролаз заключается в том, что радикал аминокислоты, по соседству с которой гидролизуется пептидная связь, служит для образования фермент-субстратного комплекса.
Пептид-гидролазы, катализирующие гидролиз пептидов до свободных аминокислот, могут отщеплять последние от пептида, начиная либо с аминокислоты, обладающей свободной NН2-группой, либо с аминокислоты, имеющей свободную COOH-группу. В первом случае их называют аминопептидазами (а-аминоацилпептид-гидролазы), во втором — карбоксипептидазами (пептидиламиноацидо-гидролазы).
Схема, поясняющая действие амино- и карбоксипептидаз, а также некоторых эндопептидаз, приведена на рис. 56.
Некоторые амино- и карбоксипептидазы обладают специфичностью действия, т. е. отщепляют строго определенные N- или С-концевые аминокислоты. Третий подподкласс пептидаз представлен дипептид-гидролазами, или дипептидазами. Их известно около десяти. Они завершают гидролиз белка. Недавно из состава пептидаз вычленено еще два подподкласса: дипептидилпептидгидролазы, отщепляющие от N-конца полипептида дипептид, и пептидилдипептид-гидролазы, отщепляющие дипептид с С-конца.
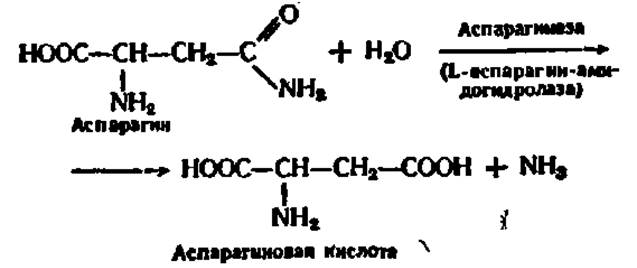
Амидазы. Эти ферменты ускоряют гидролиз амидов кислот. Из них важную роль в биохимических процессах в организме играют уреаза, аспарагиназа и глутаминаза.
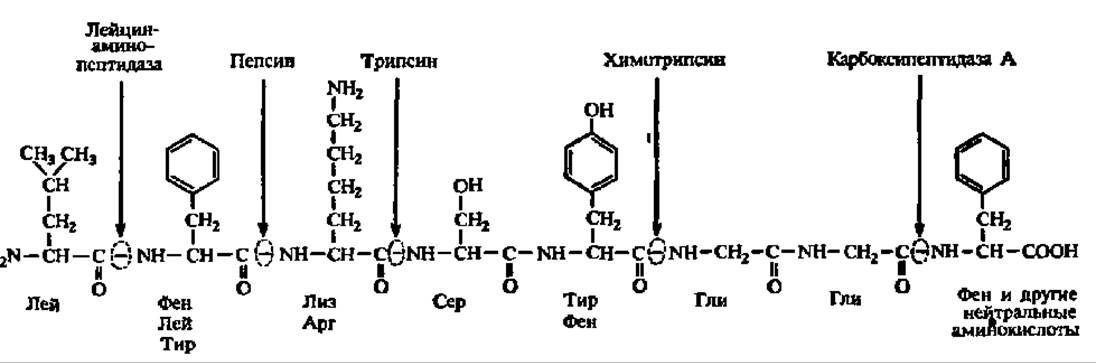
Рис. 56. Точки приложения действия протеолитических ферментов на пептидные связи в белковой молекуле
Уреаза была одним из первых белков-ферментов, полученным в кристаллическом состоянии (Д. Самнер, 1926). Это однокомпонентный фермент (М = 480 000); молекула его глобулярна и состоит из 8 равных субъединиц. Уреаза ускоряет гидролиз мочевины до NH3 и СО2.
Аспарагиназа и глутаминаза ускоряют гидролиз амидов дикарбоновых аминокислот — аспарагиновой и глутаминовой, например:
К гидролазам, действующим на С—N-связи, отличающиеся от пептидных, кроме амидаз относятся ферменты, катализирующие гидролиз С—N-связей в линейных амидинах. К их числу принадлежит аргиназа. При посредстве аргиназы аминокислота аргинин гидролизуется на орнитин и мочевину:
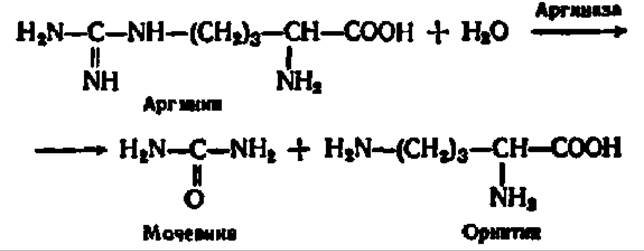
Так как в процессе гидролиза аргинина отщепляется мочевина, то систематическое название аргиназы — L-аргинин-уреогидролаза. Эта реакция широко представлена в природе, являясь заключительной стадией биосинтеза мочевины — одного из конечных продуктов распада азотсодержащих веществ. Для аргиназы, как и для уреазы, характерна абсолютная специфичность действия.
4. Лиазы. К классу лиаз относятся ферменты, ускоряющие негидролитические реакции распада органических соединений по связям С—С; С—N; С—О и т. д. При этом замыкаются двойные связи и выделяются такие простейшие продукты, как СО2, Н2О, NH3 и т. п. Некоторые из этих реакций обратимы, и соответствующие ферменты в подходящих условиях катализируют реакции не только распада, но и синтеза. Таким образом, название этого класса ферментов не всегда соответствует содержанию тех процессов, которые ими ускоряются.
Одной из важнейших групп ферментов этого класса являются углерод-углерод-лиазы (С—С-лиазы). Среди них особое значение имеют карбоксилиазы (декарбоксилазы) и альдегид-лиазы.
В природе широко распространены декарбоксилазы кетокислот и аминокислот, катализирующие реакции по следующим схемам:
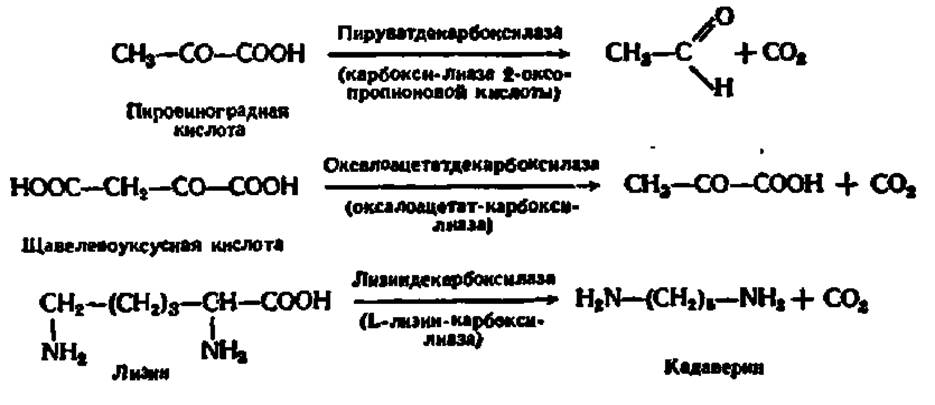
Эти ферменты двухкомпоненты, простетическими группами их во многих случаях являются фосфорные эфиры водорастворимых витаминов: тиамина (B1) — в карбокси-лиазах кетокислот и пиридоксаля (В6) — в карбокси-лиазах аминокислот.
Механизм реакции декарбоксилирования кетокислот при участии декарбоксилаз с тиаминпирофосфатом в качестве кофермента будет рассмотрен в гл. IV. Механизм реакции декарбоксилирования аминокислот с помощью карбокси-лиазы с пиридоксальфосфатом в качестве кофермента очень близок к тому, который действует при переаминировании аминокислот. Здесь тоже возникает шиффово основание, в котором электронная плотность у а-углеродного атома аминокислоты резко ослаблена. Вследствие этого сильно ослабляется связь этого углеродного атома с карбоксильной группой, и последняя легко отщепляется.
Характерным представителем альдегид-лиаз является альдолаза, катализирующая обратимую реакцию расщепления фруктозо-1,6-дифосфата до фосфотриоз:
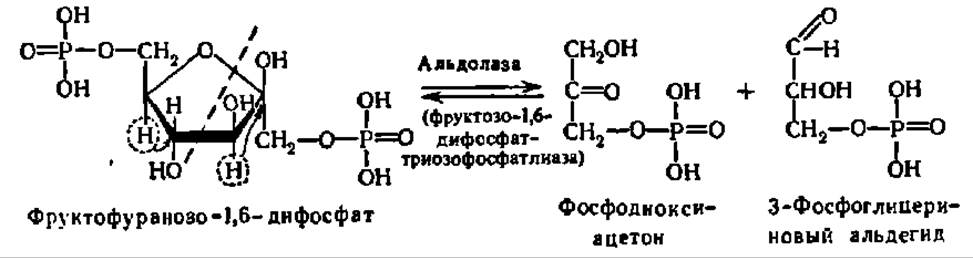
Эта реакция занимает центральное место в преобразовании углеводов. Аналогично альдолазе действуют другие альдегид-лиазы.
Другую важную группу лиаз составляют углерод — кислород лиазы (гидро-лиазы), ускоряющие реакции гидратирования и дегидратирования органических соединений. В качестве представителя гидро-лиаз приведем фумаратгидратазу:

Реакции гидратирования и дегидратирования постоянно идут при распаде и синтезе углеводов и высших жирных кислот, поэтому гидратазы играют большую роль в жизнедеятельности организмов.
Примером углерод — азот лиаз может служить аспартат — аммнак-лиаза, ускоряющая реакцию прямого дезаминирования аспарагиновой кислоты:
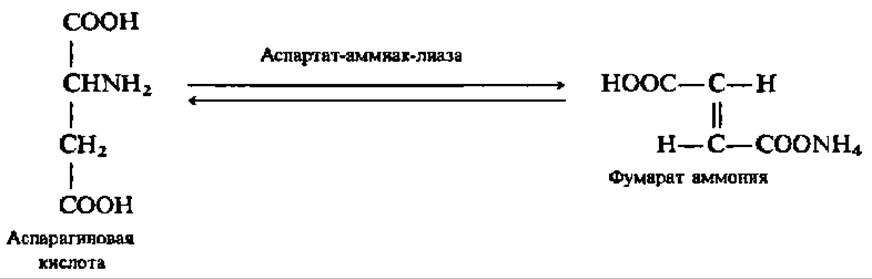
Этот фермент характерен для бактерий и ряда растений.
Некоторые лиазы ускоряют реакции не только распада, но и синтеза. Например, из дрожжей выделена L-серин-гидро-лиаза, отщепляющая от серина воду и присоединяющая сероводород, в результате чего синтезируется аминокислота — цистеин:

Чтобы отличить такие лиазы от ферментов класса лигаз (которые ускоряют реакции только синтеза и именуются в связи с этим синтетазами), их называют также синтазами.
5. Изомеразы. Ферменты, относящиеся к этому немногочисленному (около 90 индивидуальных ферментов) классу, ускоряют геометрические или структурные изменения в пределах одной молекулы. Эти изменения могут состоять во внутримолекулярном переносе водорода, фосфатных и ацильных групп, в изменении пространственного расположения атомных группировок, в перемещении двойных связей и т. п.
Важнейшими изомеразами являются триозофосфатизомераза, фосфоглицерат-фосфомутаза, альдозомутаротаза и изопентенил-пирофосфатизомераза.
Триозофосфатизомераза ускоряет перенос атомов Н в процессе превращения 3-фосфоглицеринового альдегида в фосфодиоксиацетон и обратно:
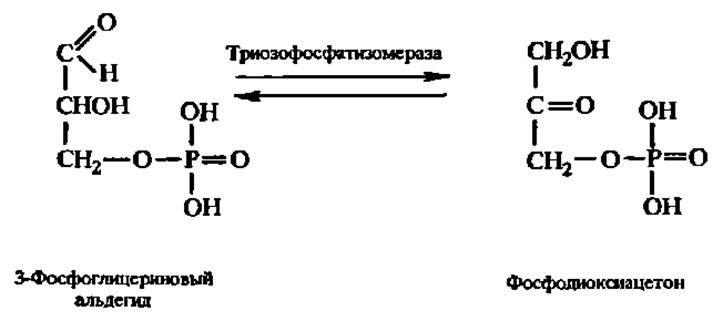
Превращение, вероятно, идет через общую эндиольную форму. Фосфоглицерат-фосфомутаза обеспечивает достаточную скорость превращения 2-фосфоглицериновой кислоты в 3-фосфоглицериновую кислоту и обратно:
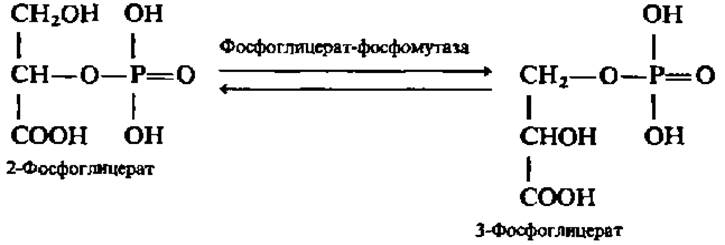
Оба процесса имеют громадное значение в органическом мире, так как представляют важнейшие стадии распада и синтеза углеводов.
Мутаротаза является представителем стереоизомераз, она ускоряет реакцию превращения a-D-глюкопиранозы в ß-D-глюкопиранозу:
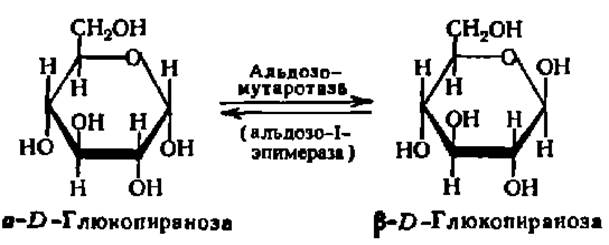
Ферменты этого типа (стереоизомеразы) обеспечивают, в частности, взаимопревращения многих пространственных изомеров моносахаридов, и этот путь является иногда единственным для синтеза некоторых из них в природе. К стереоизомеразам относятся также цис-трансизомеразы, например ретинол-изомераза, переводящая транс-ретинол в цис-ретинол (см. гл. IV).
Изопентенилпирофосфат-изомераза катализирует реакцию перестройки изопентенилпирофосфата в диметилаллилпирофосфат, что связано с перемещением двойной связи из 3-го во 2-е положение:
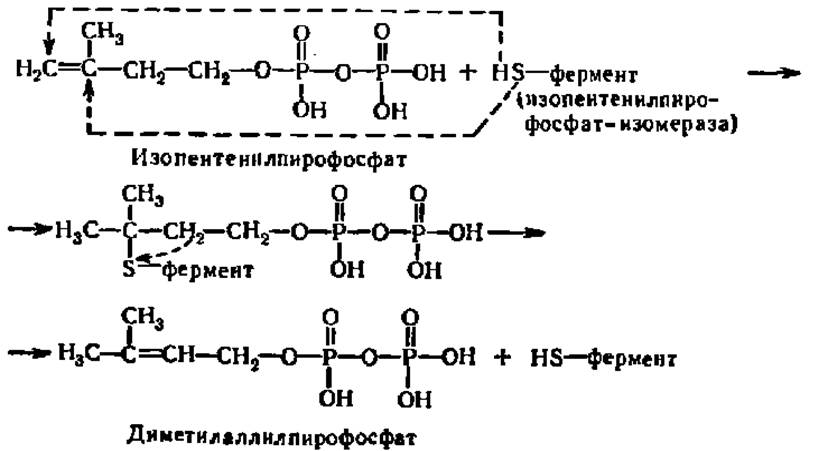
Изопентенилпирофосфат-изомераза содержит свободные сульфгидрильные группы, вероятно, в виде радикалов цис в белковой молекуле. Именно благодаря им обеспечивается указанная выше реакция, имеющая огромное значение для синтеза полиизопреноидов и стеролов.
6. Лигазы (синтетазы). Характерные черты действия ферментов этого класса выявлены совсем недавно в связи со значительными успехами в изучении механизма синтеза жиров, белков и углеводов. Оказалось, что старые представления об образовании этих соединений, согласно которым они возникают при обращении реакций гидролиза, не соответствуют действительности. Пути их синтеза принципиально иные.
Главная их особенность — сопряженность синтеза с распадом веществ, способных поставлять энергию для осуществления биосинтетического процесса. Одним из таких природных соединений является АТФ. При отрыве от ее молекулы в присутствии лигаз одного или двух концевых остатков фосфорной кислоты выделяется большое количество энергии, используемой для активирования реагирующих веществ. Лигазы же каталитически ускоряют синтез органических соединений из активированных за счет распада АТФ исходных продуктов. Таким образом, к лигазам относятся ферменты, катализирующие соединение друг с другом двух молекул, сопряженное с гидролизом пирофосфатной связи в молекуле АТФ или иного нуклеозидтрифосфата.
Механизм действия лигаз изучен еще недостаточно, но, несомненно, он весьма сложен. В ряде случаев доказано, что одно из участвующих в основной реакции веществ сначала дает промежуточное соединение с фрагментом распадающейся молекулы АТФ, а вслед за этим указанный промежуточный продукт взаимодействует со вторым партнером основной химической реакции с образованием конечного продукта.
В качестве примера действия лигазы можно привести синтез пантотеновой кислоты из а, у-диокси-ß, ß-диметилмасляной кислоты и ß-аланина:
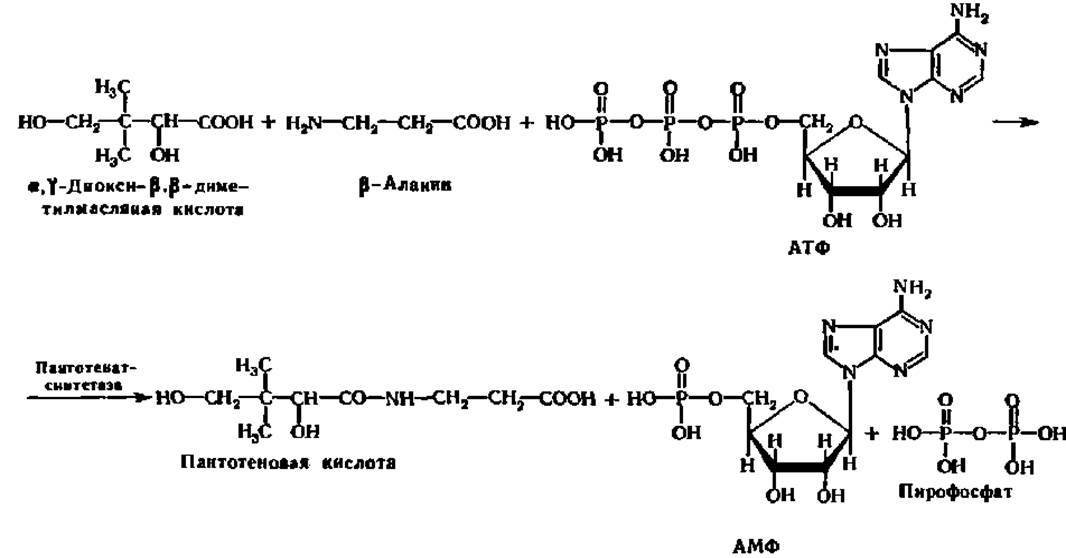
Пантотеиатсинтетаэа, как следует из приведенного уравнения, относится к группе лигаз, ускоряющих реакции синтеза С—N-связей. Эта группа лигаз насчитывает около 40 представителей. В настоящее время изучено более 75 различных лигаз, обеспечивающих важнейшие синтетические процессы в животной и растительной клетках.
Кроме ускорения реакций синтеза С—N-связей, в частности пептидных, лигазы катализируют образование связей С—С, С—О и С—S. К группе лигаз, образующих С—С-связи, относятся карбоксилазы. Они обеспечивают карбоксилирование ряда соединений, в результате чего происходит удлинение углеродных цепей. Одной из важнейших карбоксилаз является пируваткарбоксилаза, ускоряющая реакцию образования щавелевоуксусной кислоты из пировиноградной кислоты и оксида углерода (IV):
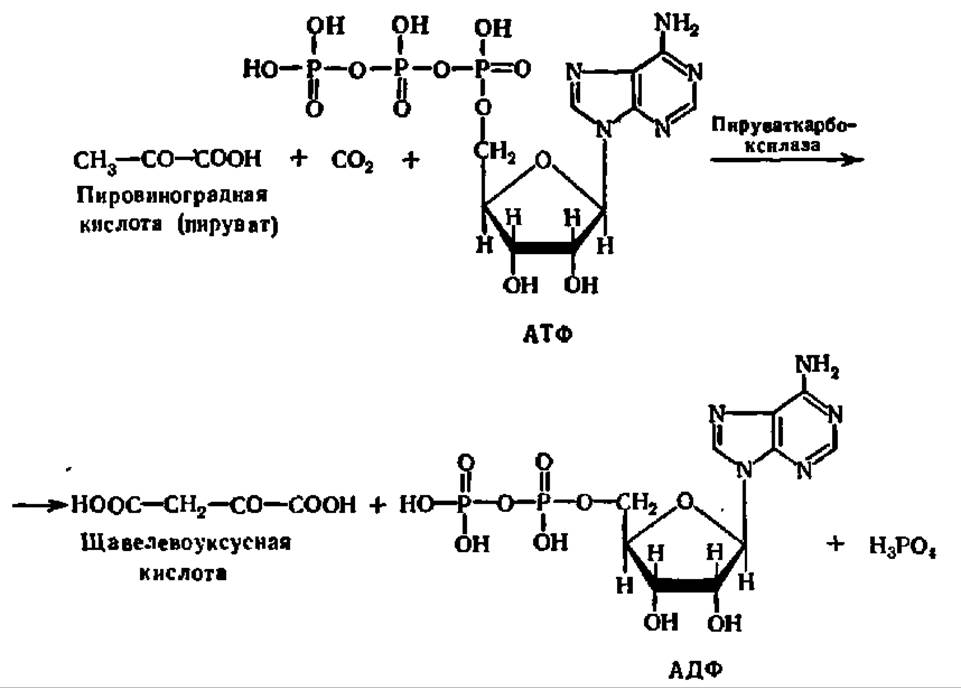
Эта реакция имеет исключительное значение в обмене веществ, обеспечивая взаимосвязь обмена углеводов и белков и акцептирование СО2.
Лигазам, катализирующим синтез С—О-связей, принадлежит важнейшая роль в биосинтезе белков, так как они ускоряют реакции активирования аминокислот перед вступлением последних в пептидную связь. Одной из простейших реакций этого типа является образование аминоациладенилатов:
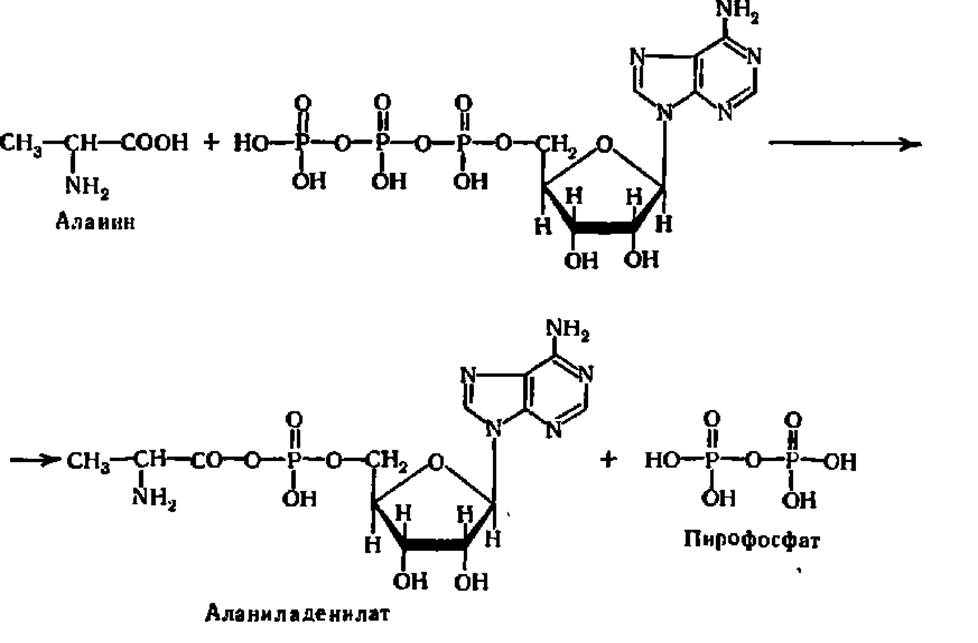
С аминоациладенилатов аминокислоты передаются на тРНК, образуя аминоацил-тРНК, используемые непосредственно при синтезе полипептидов. Эти процессы будут детально рассмотрены в гл. VII.
Лигазы, кроме того, осуществляют ускорение реакций образования С—S-связей, являясь ацил-коэизим А-синтетазами. В качестве примера действия ацил-коэнзим А-синтетазы можно привести образование ацетил-коэнзима А из уксусной кислоты и коэнзима А (см. формулу на с. 163), протекающее сопряженно с распадом АТФ:
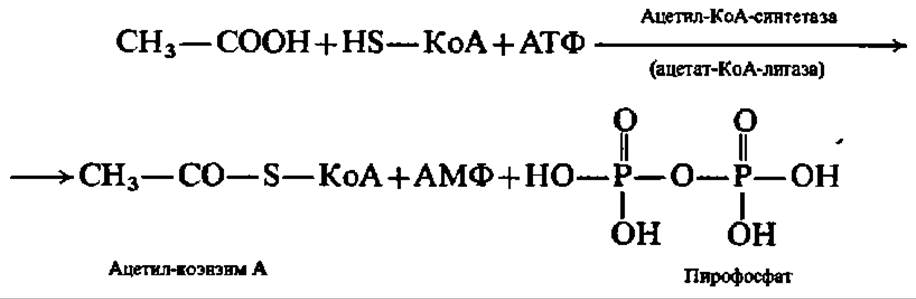
Ацетил-КоА служит коферментом в реакциях трансацилирования, поэтому действие ферментов этих двух групп — лигаз и ацилтрансфераз — в живых системах тесно увязано друг с другом. Аналогичная взаимозависимость характерна и для многих других ферментов.