Основы биохимии - Филиппович Ю. Б. 1999
Обмен белков
Распад белков и аминокислот
Пути распада белков. Главный, но возможно не единственный путь распада белков в организме—гидролиз. Гидролитический распад белков протекает в любой клетке организма в основном в специальных субклеточных элементах — лизосомах, где сосредоточены гидролитические ферменты и где осуществляется деструкция высокомолекулярных веществ до низкомолекулярных метаболитов. Вместе с тем определенная часть ферментов, ускоряющих распад белков, есть в цитозоле клетки, а некоторые из них секретируются, обеспечивая внеклеточное переваривание белков. В ряде органов и тканей (пищеварительная система животных, запасающие органы растений и т. п.) гидролиз белков осуществляется с огромной интенсивностью и в большом масштабе. Так, в печени крысы ежедневно распадается около 40% белков, а время полужизни белков важнейших субклеточных структур (ядро, рибосомы, митохондрии) и цитозоля составляет около 5 суток, хотя есть и более короткоживущие (сутки и менее) и более длительно существующие (до двухтрех месяцев) белки и ферменты.
В последние годы выяснено, что время полужизни белка в клетке детерминировано природой его N-концевой аминокислоты. Если она легко соединяется с небольшим (М = 8500 Да, 74 аминокислотных остатка, первичная структура установлена) белком — убиквитином по АТФ-зависимой реакции, то такой убиквитинированный белок атакуется протеиназами и разрушается. Наиболее подвержены убиквитинированию (перечислены в порядке убывания) арг, лиз, асп, асн, три, лей, фен, гис, глу, тир, глн, иле. N-концевые аминокислоты, менее подверженные реакции с убиквитином (мет, сер, ала, тре, вал, гли, цис), относят к стабилизирующим гидролитический распад белков. Подсчитано, например, что время полужизни цитоплазматических белков, имеющих в качестве N-концевой аминокислоты арг, составляет 2 мин, асп, лиз, лей и фен — 3 мин, про — 7 мин, глн и тир — 10 мин, глу и иле —30 мин, гли, ала, сер, вал, тре и мет — 20 ч.
Гидролиз белков может быть частичным (до пептидов) и полным (до аминокислот). При частичном (неполном) гидролизе в белковой молекуле распадаются лишь некоторые пептидные связи, как правило, по соседству со строго определенными аминокислотными радикалами. Этот процесс ускоряется специфическими ферментами—протеиназами (пептидил-пептидгидролазами). В свою очередь, пептиды гидролизуются до аминокислот, что происходит при участии ряда пептидаз. И химизм процесса гидролиза белков, и соответствующие ферменты, его ускоряющие, охарактеризованы ранее (см. гл. ІІІ).
Таким образом, в результате деятельности разнообразных пептидгидролаз (протеиназы и пептидазы) из белков в процессе их гидролиза сначала образуются сложные смеси различных пептидов, а затем смесь свободных белковых аминокислот. Последние являются конечным продуктом гидролиза белков. Механизм действия пептидгидролаз в ряде случаев изучен детально. Это касается, например, механизма действия химотрипсина,- упрощенная схема которого представлена на рис. 89.
Роль протеиназ в организме не сводится лишь к фрагментированию белковых молекул до пептидов для обеспечения дальнейшего гидролиза последних до свободных аминокислот. В последнее время все большее значение придают именно способности протеиназ селективно расщеплять полипептидные цепи, в результате чего из белковых предшественников возникают функционально активные белки и многие биологически активные пептиды, в том числе гормоны, рилизинг-факторы, психотропные пептиды и т. п. Это имеет огромное значение для регуляции обмена веществ. Протеолиз выступает как особая форма биологического контроля, однонаправленно обеспечивающего инициацию определенного физиологического процесса.
В последние годы привлекли внимание протеиназы, действие которых активируется Са2+. Их называют кальпаинами (М = 110 к Да, 2 субъединицы: каталитическая — 80 кДа и регуляторная — 30 кДа). Они расщепляют белки по границам их доменов, связывая минеральный обмен с регуляцией метаболизма. Их действие ингибируется калыюстатином.
Активный транспорт аминокислот через биологические мембраны. Свободные аминокислоты, возникающие в результате гидролитического распада белков, используются в основном для ресинтеза белковых тел и лишь некоторая их часть подвергается дальнейшей деструкции. Кроме того, содержание свободных аминокислот в клетке постоянно пополняется за счет их синтеза de novo, охватывающего весь спектр протеиногенных аминокислот у аутотрофов и заменимых аминокислот у гетеротрофов. Естественно, что существуют системы транспорта аминокислот через мембраны, обеспечивающие их перенос как через внешнюю клеточную мембрану, так и через систему внутриклеточных мембран, что обеспечивает их участие в обменных процессах, развертывающихся в компартментах клетки.
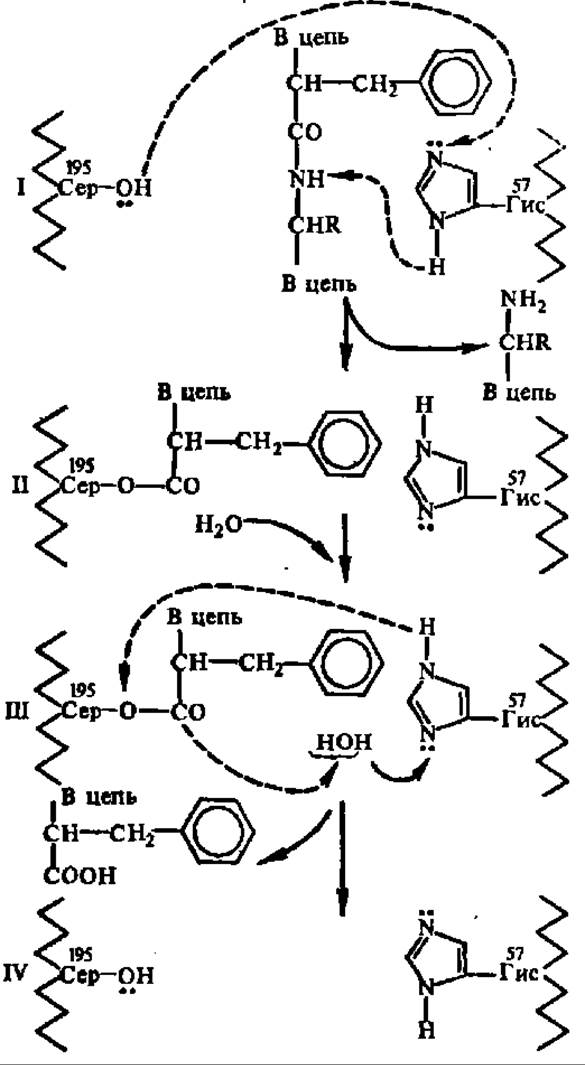
Рис. 89. Упрощенная схема гидролиза пептидной связи в активном центре химотрипсина
Активный центр фермента содержит остатки серина (195-е положение в полипептидной цепи) и гистидина (57-е положение в той же полипептидной цепи). Фрагменты единой полипептидной цеди молекулы фермента условно показаны слева и справа изогнутой линией, в их составе отмечены остатки сер я гис с их функциональными группами. В активном центре фермента точно против остатков сер и гис размешается пептидная связь гидролизуемого белка, образованная остатком феи (или тир, три, лей), благодаря контакту радикала фен (иля тир, три, лей) с адсорбционным (субстратным) центром фермента, который на рисунке не показан. Вследствие того, что электронная пара кислорода гидроксильной группы радикала сер акцентируется карбонильной группой пептидной связи, создаются условия для миграции протона от гидроксила сер к имидазольному радикалу остатка гис и для ацилирования радикала сер с одновременным разрывом пептидной связи. Одновременно протон от имидазольного ядра радикала гис перемещается к NH-гpyппe деструктируемой пептидной связи (I). После ацилирования радикала сер (II) в активный центр фермента входят молекула воды, инициирующая распад ацильного производного фермента (III) и восстановление исходного состояния активного центра (IV), готового принять новую молекулу субстрата или атаковать пептидную связь, образованную остатками фен, тир. пут или лей в том же субстрате
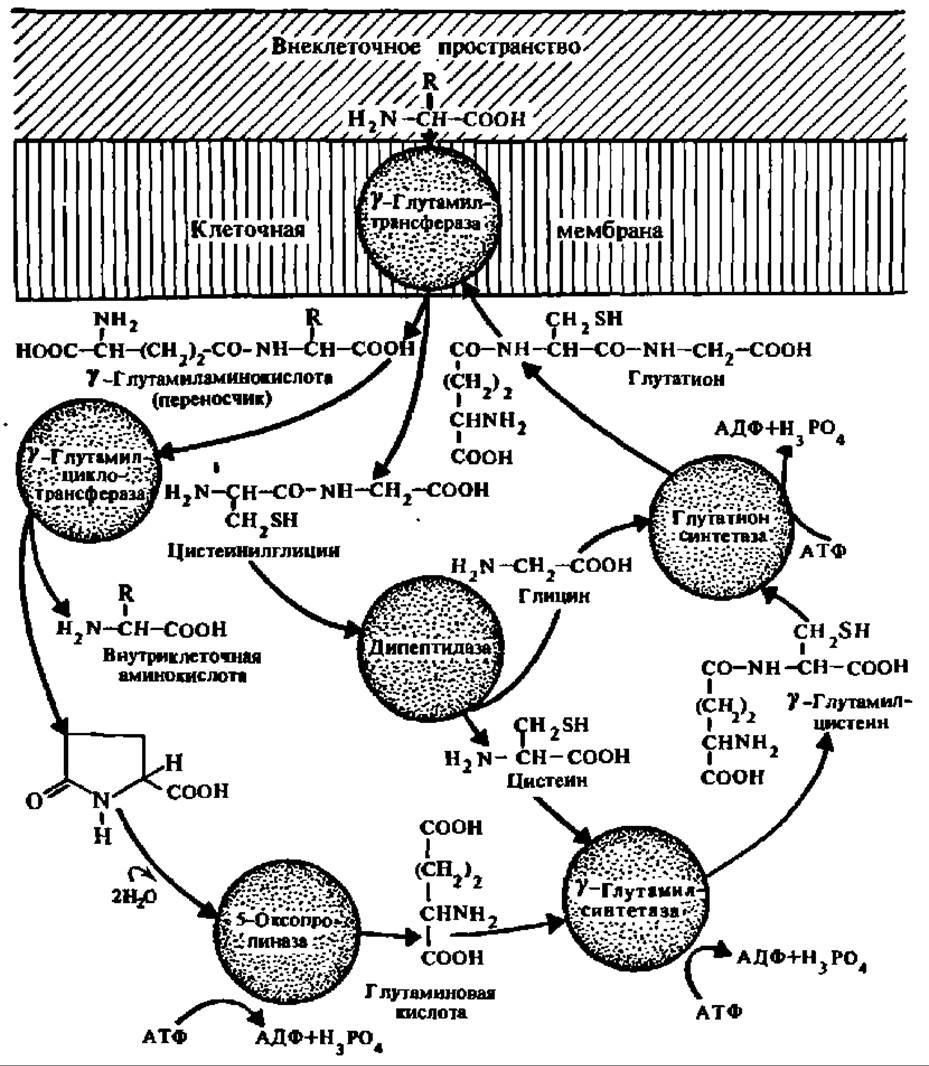
Рис. 90. у-Глутамилтрансферазный цикл
у-Глутамилтрансфераза встроена в клеточную мембрану и осуществляет транслокацию аминокислот из внеклеточного пространства за счет реакции транслептидирования остатка у-глутаминовой кислоты с глутатиона или другого у-глутамилпептида на транспортируемую аминокислоту и переноса возникшего переносчика, а именно у-глутамиламниокислотного производного, во внутриклеточное (или внутримембранное) пространство. Здесь благодаря действию у-глутамилциклотрансферазы переносчик распадается на свободную аминокислоту, которая таким образом оказывается перенесенной через мембрану, и пироглутаминовую кислоту, образование которой практически напело сдвигает реакцию распада дипептида — переносчика вправо. В результате ряда ферментативных процессов (правая часть рисунка) происходит ресинтез глутатиона (или другого у-глутамилпептида, если он участвует в переносе аминокислот), и цикл может повториться снова
Проблему активного переноса аминокислот через биологические мембраны интенсивно разрабатывали многие исследователи. А. Майстер (1973) предложил гипотезу переноса аминокислот через мембраны при посредстве у-глутамилтрансферазного цикла, сущность которой ясна из рассмотрения рис. 90. Согласно этой гипотезе, центральную роль в данном процессе играет фермент у-глутамилтрансфераза. Естественно, что транслокация аминокислот через биологические мембраны осуществляется также белками-переносчиками (см. рис. 43). Такие системы переноса изучены для гистидина, лейцина, изолейцина и валина.
Превращения аминокислот. Соотношения аминокислот в распадающихся белках и новообразуемых за их счет протеинах, как правило, различны. Поэтому известная доля свободных аминокислот, возникших при гидролизе белков и пептидов, обязательно должна быть преобразована либо в другие аминокислоты, либо в более простые соединения, выводимые из организма. Все это осуществляется в результате процессов, которые можно объединить общим названием — превращения аминокислот.
Известны три типа реакций аминокислот в организме: по а-аминогруппе, карбоксильной группе и радикалу аминокислоты.
Реакции по а-аминогруппе однотипны у всех аминокислот, это в основном реакции дезаминирования и переаминирования. Столь же однообразен набор химических процессов по карбоксильной группе аминокислот: это главным образом декарбоксилирование и образование аминоациладенилатов. В отличие от первых двух типов превращений аминокислот преобразования в радикалах аминокислот исключительно разнообразны, многочисленны и, как правило, уникальны для каждой отдельной аминокислоты. Наконец, есть тип превращений аминокислот, который состоит в образовании пептидной связи между а-аминогруппой одной аминокислоты и карбоксильной группой другой. Он осуществляется сложным путем и приводит к синтезу пептидов и белков. Здесь рассматриваются лишь первые три типа превращений аминокислот, а синтез из них пептидов и белков — ниже, в этой же главе.
Реакции по аминогруппе. Наиболее распространенной и важной реакцией аминокислот по а-аминогруппе является дезаминирование. Оно может идти четырьмя путями:
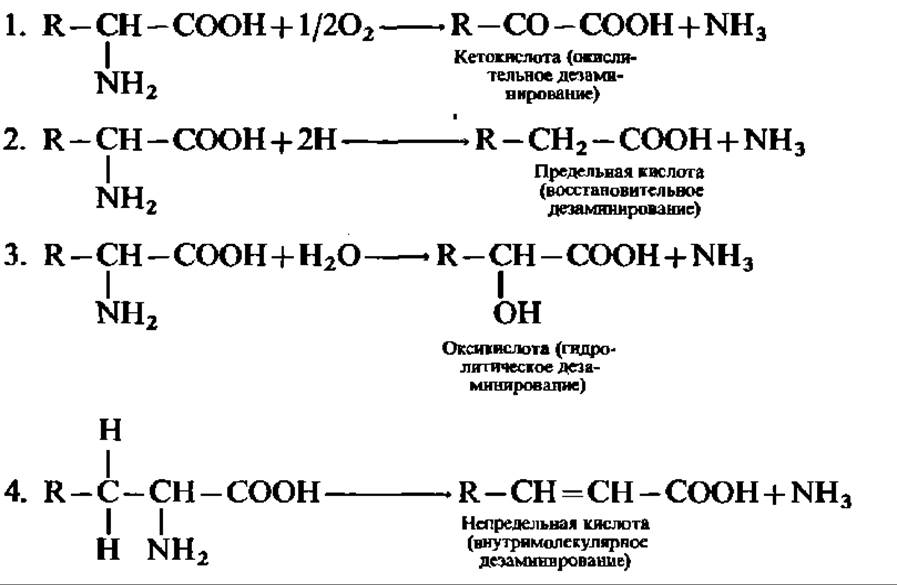
Все перечисленные реакции действительно осуществляются в организмах и каждая из них ускоряется специфическим ферментом; однако распространение их в природе совершенно различно: очень широко распространена 1-я реакция, а остальные три встречаются крайне редко, лишь у отдельных групп организмов.
Так как преобладающим является окислительное дезаминирование, рассмотрим его подробнее. Процесс этот осуществляется в две стадии. Сначала аминокислота окисляется в иминокислоту при участии специфической дегидрогеназы с НАД+ или НАДФ+ в качестве кофермента и акцептора водорода. Затем иминокислота спонтанно гидролизуется на кетокислоту и аммиак:
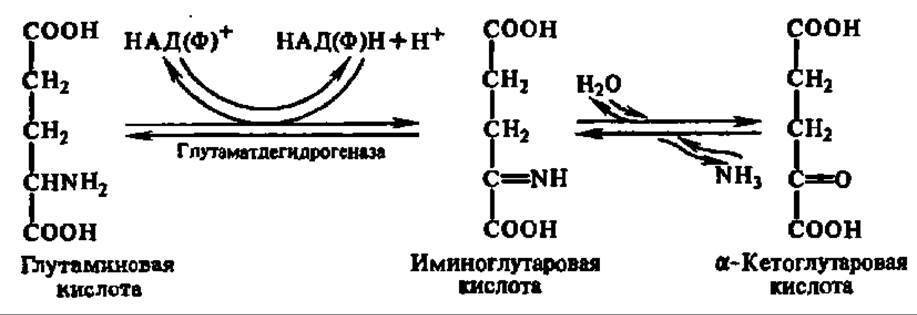
Обе реакции обратимы, и таким путем из а-кетоглутаровой кислоты и аммиака в организме образуется глутаминовая кислота.
В некоторых случаях дегидрогеназы аминокислот представлены флавопротеинами. Так, из грены шелкопряда выделена дегидрогеназа а-аминокислот, отличающаяся очень высокой активностью и несущая флавиновую группировку в составе кофермента.
Полная эпимолекула глутаматдегидрогеназы (М = 336 000) составлена из 6 субъединиц с М = 56 000 (см. рис. 46). Каждый из протомеров имеет центры связывания субстрата, кофермента и эффекторов (АДФ и ГДФ активируют, а АТФ, ГТФ и пиридоксальфосфат ингибируют фермент). Первичная структура субъединиц ряда глутаматдегидрогеназ, выделенных из разных объектов, отличается высокой степенью гомологии.
Дегидрогеназы L-аминокислот (в отличие от дегидрогеназ D-аминокислот) в тканях животных и растений представлены слабо, и только дегидрогеназа L-глутаминовой кислоты проявляет себя исключительно ярко. Поэтому допускают, что большинство L-аминокислот дезаминируется в организме путем переаминирования с а-кетоглутаровой кислотой по уравнению:
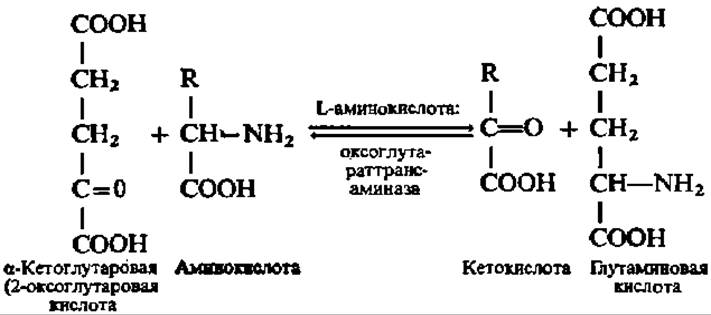
Вслед за этим глутаминовая кислота претерпевает окислительное дезаминирование, а выделяющаяся при этом а-кетоглутаровая кислота снова вовлекается в реакцию переаминирования с а-аминокислотами.
Выделены специфические трансаминазы, ускоряющие реакцию переаминирования между почти всеми белковыми аминокислотами и а-кетоглутаровой кислотой. Механизм реакции переаминирования детально рассмотрен в гл. III. Реакция переаминирования между L-аминокислотами и а-кетоглутаровой кислотой обратима, поэтому при определенных условиях она служит для синтеза L-аминокислот из кетокислот и глутаминовой кислоты. Таким образом, реакцию переаминирования нельзя сводить только к дезаминированию аминокислот; ее роль в организме гораздо шире.
Главным продуктом дезаминирования аминокислот являются а-кетокислоты. Лишь в некоторых, особых и мало распространенных случаях в качестве конечных продуктов дезаминирования аминокислот отмечены предельные или непредельные жирные кислоты, а также оксикислоты.
Дезаминирование некоторых аминокислот идет своеобразно. Так, серосодержащие аминокислоты (цистеин и метионин) дезаминируются путем отщепления аммиака и сероводорода или метилмеркаптана (CH3SH) соответственно; оксиаминокислоты (серин и треонин) — путем отщепления аммиака и воды; гетероциклические аминокислоты —путем дегидрирования по кольцу (пролин) с дальнейшим преобразованием продукта дегидрирования и т. д. Однако и в этих случаях конечными продуктами дезаминирования остаются кетокислоты и непредельные кислоты.
Реакции по карбоксильной группе. Превращения аминокислот по СООН- группам сводятся в основном к декарбоксилированию и образованию аминоациладенилатов. Декарбоксилирование аминокислот осуществляется сравнительно легко в тканях животных и растений, но особенно широко оно представлено у микроорганизмов. Во всех случаях процесс идет по одной и той же схеме:

Простатической группой декарбоксилаз L-аминокислот служит пиридоксальфосфат, комплекс которого с различными специфическими белками дает начало всем многообразным и высокоспецифичным декарбоксилазам L-аминокислот. Выделены и изучены декарбоксилазы аспарагиновой и глутаминовой кислот, валина, лизина, аргинина, гистидина, тирозина, триптофана и ряда других аминокислот. Механизм действия их ясен из схемы, приведенной на с. 127, и уравнений реакций на с. 133.
В подавляющем большинстве случаев продуктами декарбоксилирования аминокислот являются амины. Так как они образуются в качестве продуктов жизнедеятельности и обладают высокой физиологической активностью, их называют биогенными аминами. Приведем некоторые примеры.
При декарбоксилировании гистидина возникает гистамин:
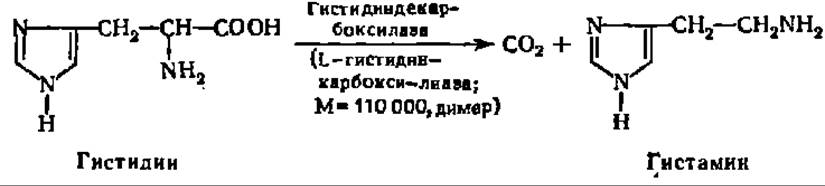
Он вызывает усиление деятельности желез внутренней секреции и снижает кровяное давление.
При декарбоксилировании тирозина и триптофана образуются соответственно тирамин и триптамин:
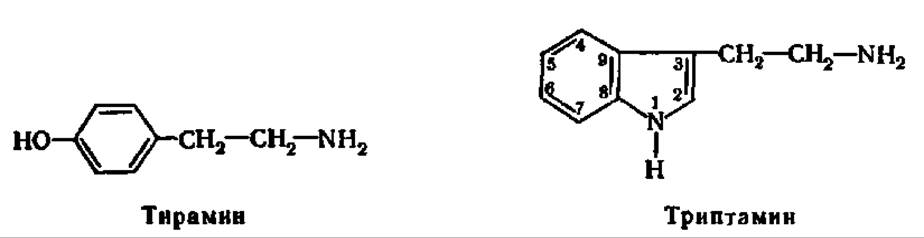
Последний легко переходит в 5-окситриптамин (серотонин) — соединение, обладающее многогранным физиологическим действием, имеющее, в частности, отношение к возникновению болевых ощущений при воспалительных процессах.
Декарбоксилирование лизина и аргинина сопровождается образованием кадаверина и агматина:
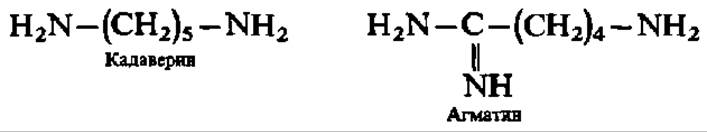
В последнее время важное значение придают тетраметилендиамину (путресцин), возникающему при декарбоксилировании аминокислоты — орнитина:

Тетраметилендиамин служит в организме исходным соединением для синтеза спермидина и спермина (см. с. 211). Оба эти вещества — полиамины, обеспечивающие наряду с диаминами определенные структурные особенности и функциональную активность рибосом.
Не только амины и диамины являются продуктами декарбоксилирования аминокислот. При декарбоксилировании глутаминовой кислоты образуется у-аминомасляная кислота:
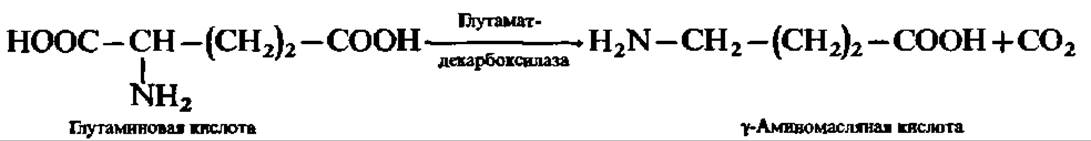
Она накапливается в мозговой ткани и представляет собой нейрогуморальный ингибитор. Аналогично этому из аспарагиновой кислоты получается ß-аланин:
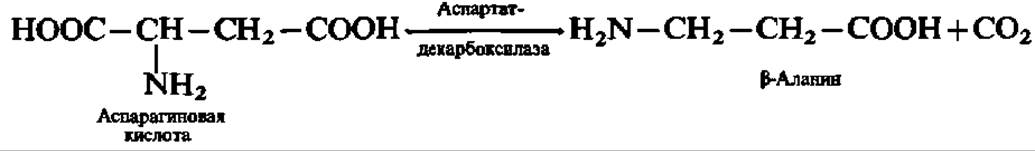
Он принимает участие в синтезе пантотеновой кислоты (см. с. 162).
Второй важной реакцией аминокислот по карбоксильной группе является образование ими аминоациладенилатов. Эта реакция была отмечена ранее при рассмотрении ферментов, относящихся к классу лигаз (см. с. 137), и будет подробно освещена ниже, в этой главе.
Превращения аминокислот, связанные с реакциями по радикалу. Напомним прежде всего, что радикалом аминокислоты принято называть ту часть ее молекулы, которая не принимает участия в формировании хребта полипептидной цепи. По своей химической природе радикалы аминокислот исключительно разнообразны, что служит материальной основой для многообразия присущих им химических реакций. Естественно, что многие из этих реакций осуществляются в процессе обмена аминокислот.
Важнейшим типом превращений аминокислот, протекающих с видоизменением радикалов, является переход одних аминокислот в другие. Благодаря этому в организме значительно усиливаются возможности для синтеза аминокислот. Приведем некоторые примеры.
При окислении фенилаланина образуется тирозин:
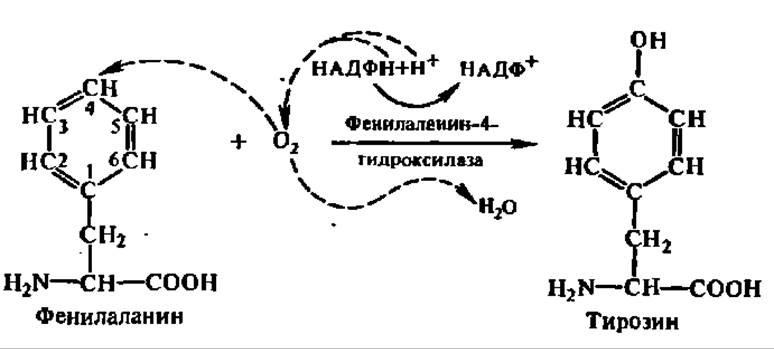
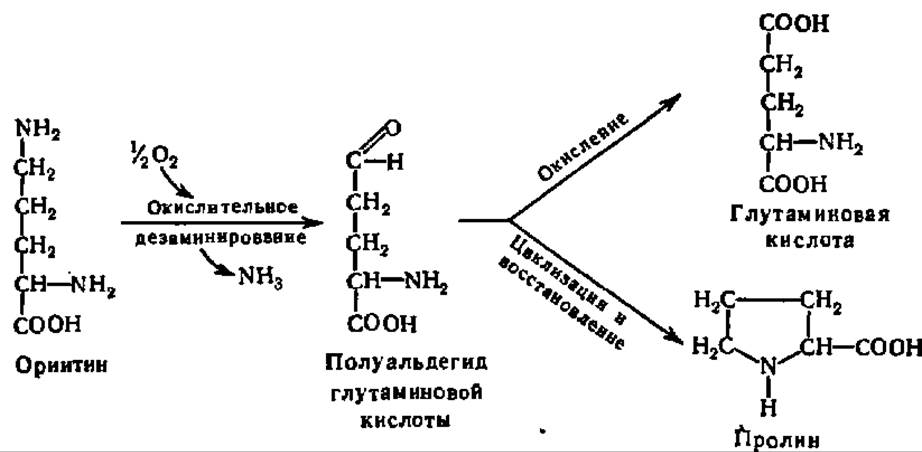
Гидролиз аргинина приводит к образованию аминокислоты — орнитина:
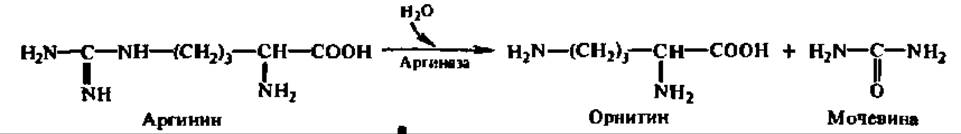
Из последнего, в свою очередь, возникает либо глутаминовая кислота, либо пролин:
Окислительно-восстановительные процессы легко протекают по серосодержащим радикалам цистеина и цистина, благодаря чему эти две аминокислоты переходят друг в друга; химизм этой реакции аналогичен окислению глутатиона (см. с. 48), а ускоряется она цистеинредуктазой.
При исчерпывающем окислении тиоловой группы цистеина последний переходит в цистеиновую кислоту, которая, декарбоксилируясь, дает начало таурину:
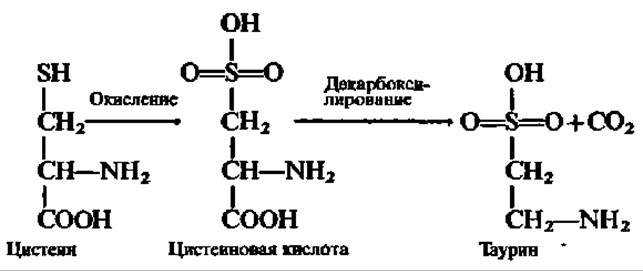
Последний, соединяясь с желчными кислотами, например холевой кислотой (см. с. 380), принимает участие во всасывании жиров.
Большое распространение имеет реакция диметилирования метионина. Она осуществляется при каталитическом воздействии метилтрансферазы. Метионин — универсальный поставщик метальных групп в реакциях трансметилирования. При этом он переходит сначала в «активный метионин», соединяясь с АТФ:
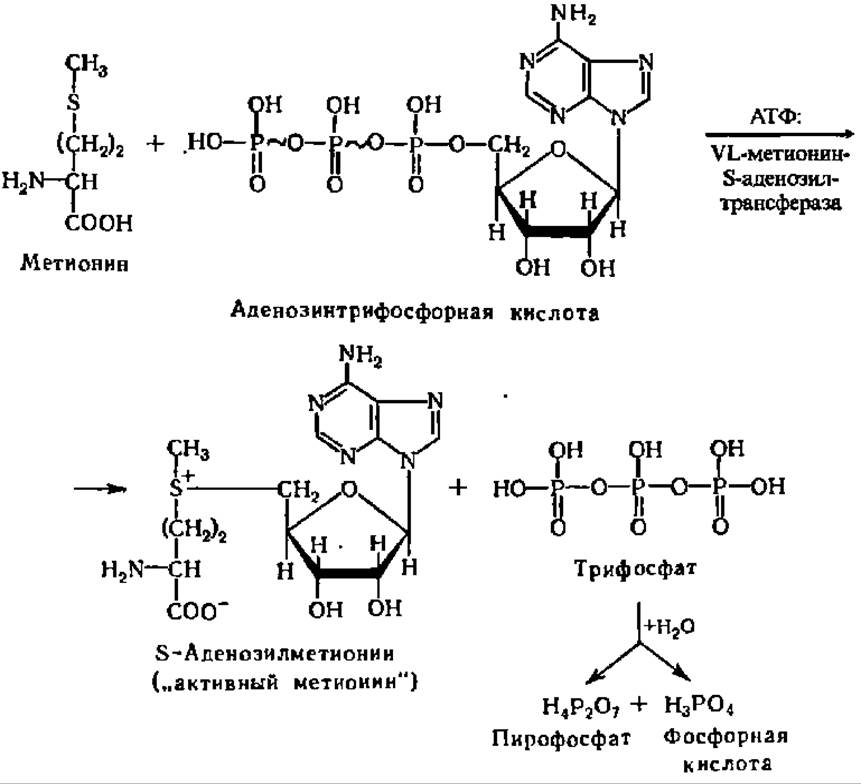
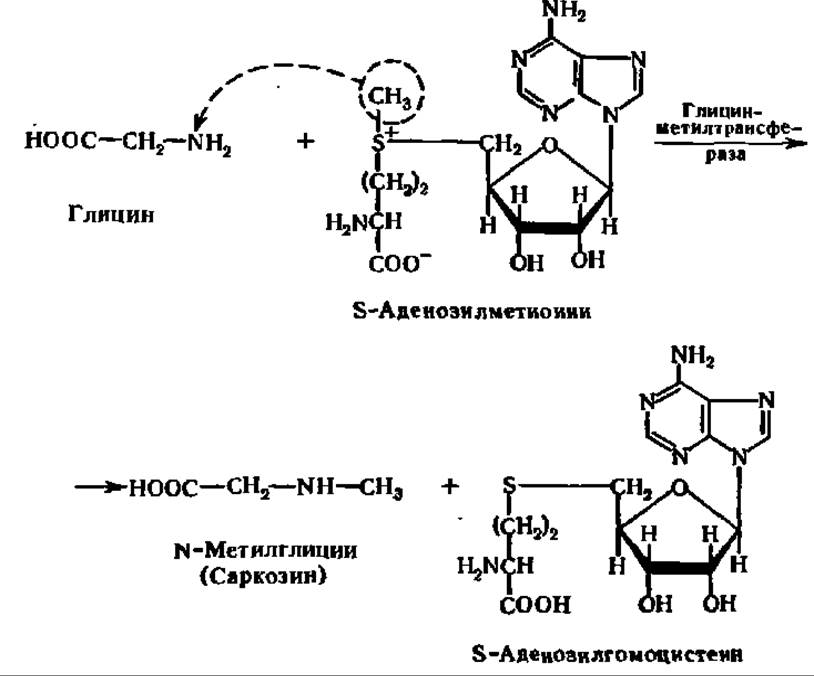
Реакция ускоряется специфическим ферментом АТФ: L-метионин — S-аденозил трансферазой (М = 100 000, оптимум pH 9,5). Далее метальная группа от S-аденозилметионина передается соединению, которое подвергается метилированию. В качестве примера приведем уравнение реакции метилирования глицина:
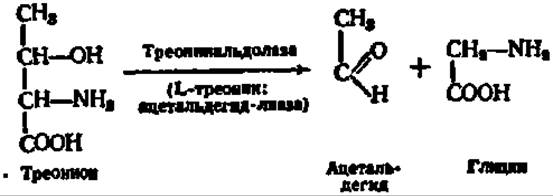
Интересная реакция осуществляется по радикалу треонина. Она состоит в отщеплении радикала в виде ацетальдегида. Эта реакция ускоряется специфическим ферментом — треонинальдолазой, открытым А. Е. Браунштейном и Г. Я. Виленкиной (1951). Простетической группой его является пиридоксальфосфат. В качестве второго продукта расщепления треонина идентифицирован глицин:
Кроме реакций, приводящих к синтезу одних аминокислот из других, по радикалам аминокислот известно много других превращений (окисление, метилирование и т. п.). Часто эти реакции сочетаются с процессами декарбоксилирования и дезаминирования аминокислот. В результате (особенно из циклических аминокислот) возникают разнообразные вещества, многие из которых обладают сильным физиологическим действием. Так, например, из тирозина образуется гормон адреналин (см. гл. XII). Триптофан служит источником образования никотиновой кислоты (витамин РР) и индолилуксусной кислоты (ростовое вещество); цистеин — меркаптуровых кислот (обезвреживание ароматических соединений); аргинин — аргининфосфата и других гуанидинфосфатов (макроэргические соединения).
Таким образом, в процессе превращений аминокислот возникает серия соединений, принимающих участие в регуляции обмена веществ в организме. Это обстоятельство еще раз подчеркивает ведущую роль белкового обмена в общем обмене веществ организма.
Конечные продукты распада аминокислот. Как было отмечено выше, в результате распада аминокислот возникают СО2, NH3, амины, кетокислоты и в ряде случаев еще достаточно сложные вещества, относящиеся к тем или иным классам органических соединений. Все они, за исключением СО2 и NH3, подвергаются в конце концов дальнейшей деструкции. Амины путем окислительного дезаминирования превращаются в карбоновые кислоты:
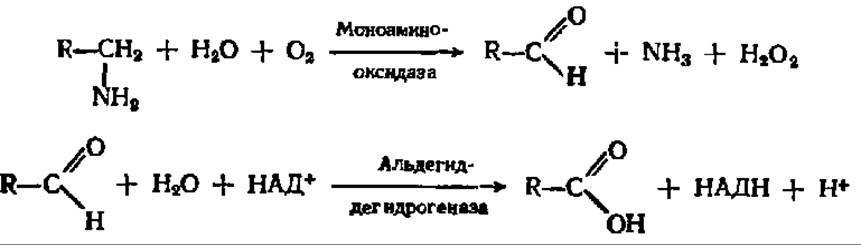
Аналогично идет реакция окислительного дезаминирования диаминов при посредстве диаминоксидазы.
Кетокислоты и карбоновые кислоты, возникающие в результате распада аминокислот, постепенно окисляются, образуя СО2 и Н2О (см. с. 356). Точно так же в СО2, NH3 и Н2О превращаются все остальные органические вещества, являющиеся продуктами распада аминокислот в организме. Таким образом, конечными продуктами распада аминокислот являются Н2О, СО2 и NH3. Вода поступает в общий метаболический фонд, оксид углерода (IV) беспрепятственно выводится из организма и лишь судьба аммиака нуждается в специальном рассмотрении.
Только, у некоторых обитателей гидросферы (медицинская пиявка, крабы, речной рак, беззубка, каракатица и др.) NH3 непосредственно или в виде солей аммония выводится в окружающую среду. У подавляющего большинства растительных и животных видов аммиак, уже в небольших концентрациях оказывающий вредное влияние на жизнедеятельность организмов, переводится в безвредные для биологических форм азотистые соединения. К их числу относятся аспарагин, глутамин и мочевина. У многих животных, особенно позвоночных, последняя служит для выведения обезвреженного аммиака.
Полагают, что аспарагиновая и глутаминовая кислоты осуществляют первичное связывание NH3 в момент его образования в клетке. Взаимодействие аммиака с этими кислотами, в результате чего возникают амиды — аспарагин и глутамин, ускоряется специфическими ферментами. Оба фермента — аспарагинсинте- таза и глутаминсинтетаза — принадлежат к классу лигаз, в частности к подклассу лигаз, ускоряющих реакции образования С—N-связей, и среди них — к подподклассу кислотоаммиачных лигаз (амидосинтетазы). Так как необходимым условием деятельности лигаз является сопряженный с реакцией синтеза процесс распада АТФ, то уравнение реакции биосинтеза аспарагина имеет такой вид:

Аналогично идет реакция биосинтеза глутамина при участии глутаминсинтетазы (L-глутаматаммиак-лигаза).
Реакции образования аспарагина и глутамина особенно широко представлены в растительном царстве. Однако и у животных эти амиды возникают не столь редко: синтез аспарагина доказан в жировом теле насекомых, а синтез глутамина — в мышцах, мозгу, печени и почках млекопитающих, а также гемолимфе насекомых. В тканях млекопитающих синтез аспарагина идет из глутамина, амидная группа которого переносится на у-карбоксильную группу аспарагиновой кислоты при участии глутаминзависимой аспарагинсинтетазы сопряженно с распадом АТФ на АМФ и пирофосфат.
Амидирование аспарагиновой и глутаминовой кислот может происходить и в том случае, если они находятся в связанном состоянии, например, в составе белковой молекулы. Как известно, радикалы аминокислот, входящих в полипептидную цепь белка, свободны и по ним легко осуществляются те или иные химические реакции. Одной из таких реакций является амидирование белков:
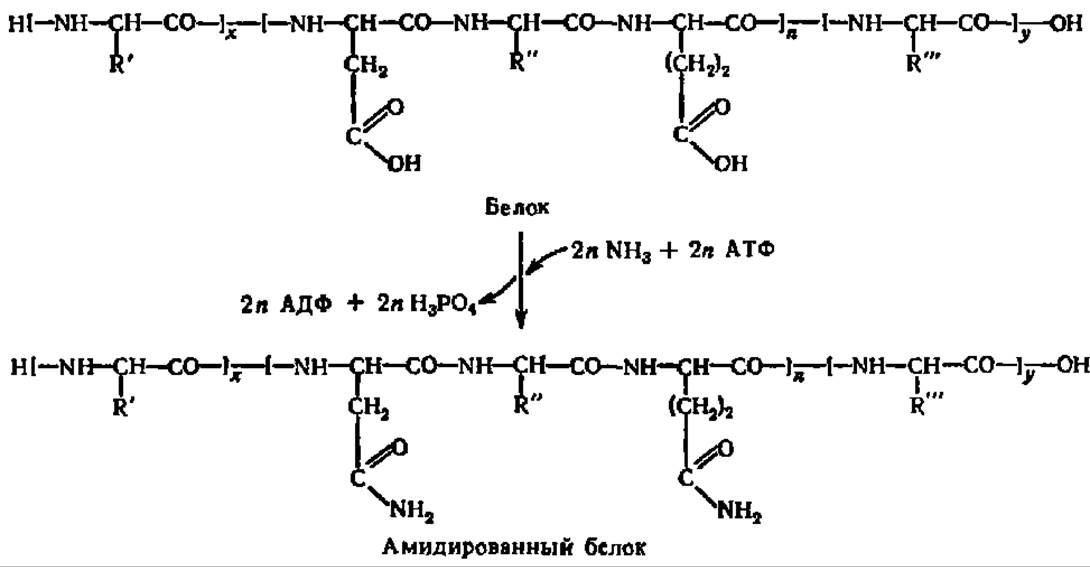
Следовательно, не только свободные аспарагиновая и глутаминовая кислоты, но и белки организма могут быть акцепторами NH3. Тем самым обеспечивается немедленное связывание аммиака в любой точке, где он возникает в результате обмена веществ. Вместе с тем амидирование белков представляет процесс посттрансляционной (см. с.301) модификации белков, в результате которой полностью завершается их синтез. Различная степень посттрансляционного амидирования является одним из источников микрогетерогенности белка.
Мочевина — основной конечный продукт белкового обмена у многих животных (дождевой червь, слизень, акула, лягушка, черепаха и все млекопитающие). Ее биосинтез у высших животных происходит в печени, на что впервые в конце прошлого столетия обратили внимание М. В. Ненцкий и И. П. Павлов. В печени найдены все необходимые для этого ферменты. У животных, не способных синтезировать мочевину (рептилии, птицы), печень не обладает соответствующим ферментативным аппаратом. Новообразование мочевины идет также в растениях. Путь ее возникновения у животных и растений одинаков и состоит в следующем. Из NH3, СО2 и АТФ при каталитическом воздействии фосфотрансферазы (карбаматкиназа) синтезируется карбамилфосфорная кислота. Механизм этой реакции рассмотрен ранее (см. с. 235). При участии другой трансферазы (орнитин-карбамилтрансфераза) карбаминовая группировка переносится от карбамилфосфата на 5-аминогруппу орнитина, который всегда присутствует в организме, так как легко возникает при гидролизе аргинина. В результате этой реакции синтезируется цитруллин:
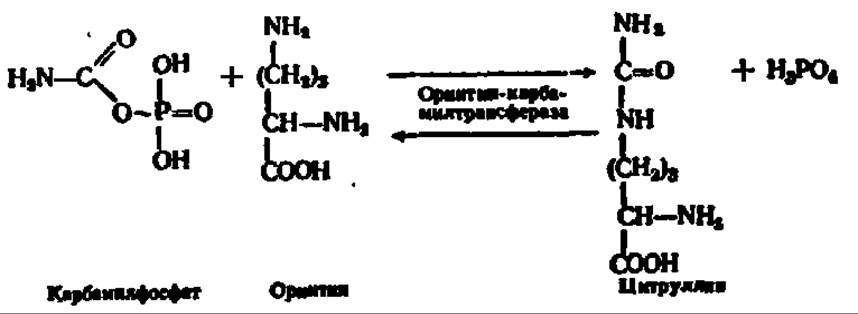
Далее в действие вступают еще два фермента, обеспечивающие введение в карбаминовую 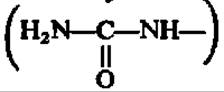 группировку цитруллина еще одного атома азота и превращение ее в гуанидиновую группировку
группировку цитруллина еще одного атома азота и превращение ее в гуанидиновую группировку 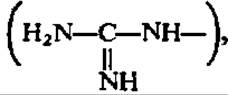 т. е. переход цитруллина в аргинин.
т. е. переход цитруллина в аргинин.
Донором аминогруппы в этом превращении служит аспарагиновая кислота, а промежуточным соединением на пути от цитруллина к аргинину — аргинин-янтарная кислота:
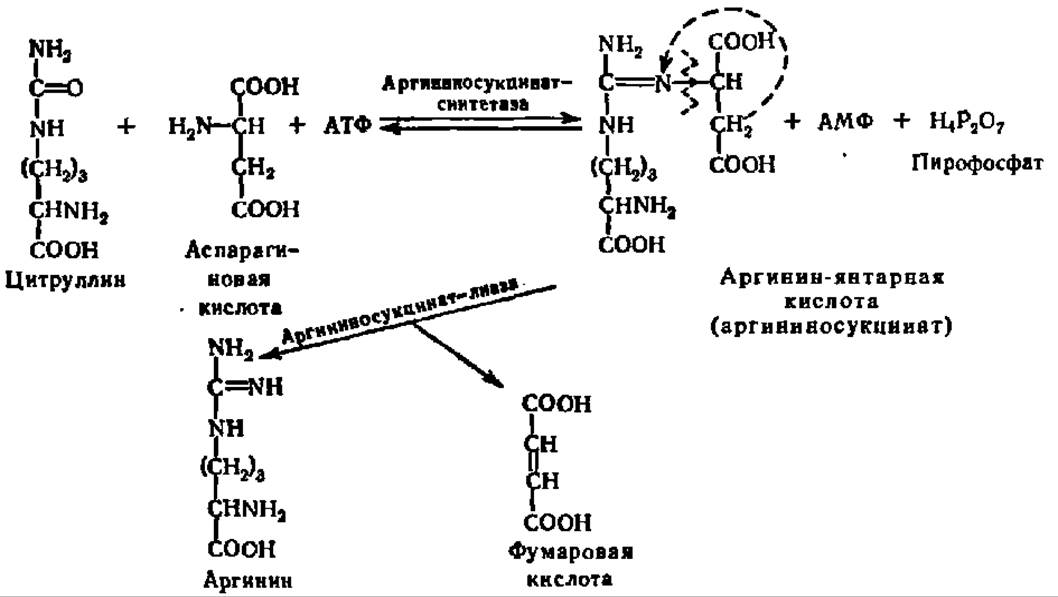
Заключительной реакцией в биосинтезе мочевины является гидролиз аргинина и образование орнитина и мочевины. Получающийся при этом орнитин вновь вступает во взаимодействие с карбамилфосфатом, и все перечисленные выше реакции повторяются снова. Поэтому совокупность указанных реакций, приводящих к образованию мочевины в качестве одного из звеньев, включающих высвобождение и вовлечение снова в процесс орнитина, получила название орнитинового цикла (рис. 91).
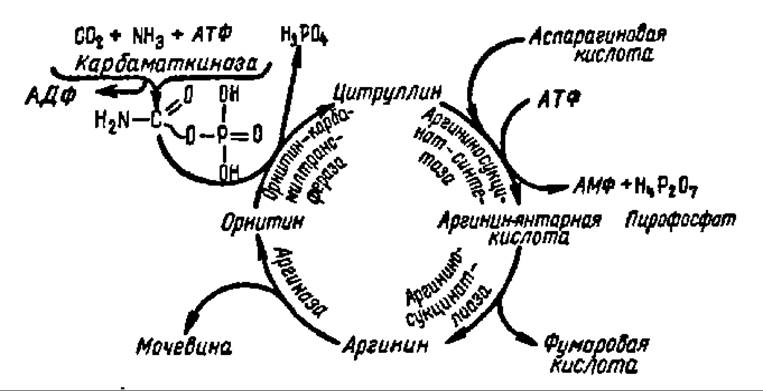
Рис. 91. Орнитиновый цикл (пояснение в тексте)
Фумаровая кислота, как мы увидим ниже, легко превращается в щавелевоуксусную кислоту, которая путем переаминирования либо аминирования переходит снова в аспарагиновую кислоту. Следовательно, с одной стороны, акцептируется молекула NH3, а с другой — возобновляются запасы аспарагиновой кислоты, участвующей в функционировании орнитинового цикла. Самое важное состоит в том, что в результате деятельности орнитинового цикла из каждых двух молекул NH3 и одной молекулы СО2, освобожденных в результате распада аминокислот (или других соединений), строится одна молекула мочевины.
Новообразование аминокислот. Выше уже было рассмотрено новообразование аминокислот в природе путем их переаминирования с кетокислотами, т. е. превращение одних аминокислот в другие. Однако в обоих случаях исходным продуктом служат уже готовые аминокислоты, которые лишь тем или иным способом видоизменяются, т. е. получаются путем вторичного синтеза из предсуществующих аминокислот. Значит, оба эти процесса не решают проблему первичного синтеза аминокислот в организме. Он осуществляется восстановительным аминированием кетокислот и прямым аминированием непредельных кислот.
Прямое аминирование непредельных кислот представляет довольно редкую реакцию и свойственно в основном бактериям и растениям. Хорошо изучено прямое аминирование фумаровой кислоты; оно ускоряется специфическим ферментом — аспартат-аммиак-лиазой и идет в соответствии со следующим уравнением:
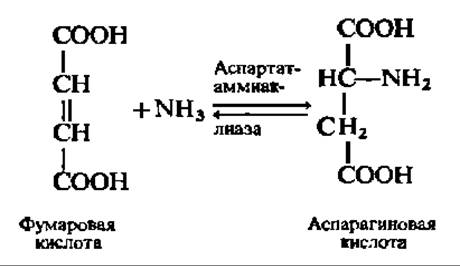
Реакция обратима и в известных условиях служит для дезаминирования аспарагиновой кислоты. Иммобилизованная аспартат-аммиак-лиаза нашла (см. с. 144) применение для промышленного получения L-аспарагиновой кислоты. Значительные количества аспарагиновой кислоты синтезируются также путем переаминирования щавелевоуксусной кислоты с глутаминовой кислотой.
Восстановительное аминирование представляет главный путь новообразования аминокислот. Эта реакция есть обращение окислительного дезаминирования аминокислот. Ее уравнение приведено на с. 265. Если его прочесть справа налево, то это и будет уравнение реакции восстановительного аминирования а-кетоглутаровой кислоты.
Другой кетокислотой, подвергающейся активно восстановительному аминированию, является пировиноградная кислота:
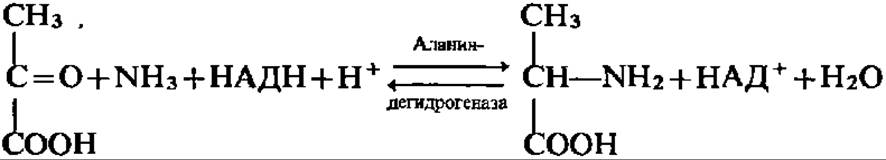
В принципе, возможно восстановительное аминирование любой кетокислоты. Однако активность всех природных дегидрогеназ аминокислот, за исключением глутамат- и аланиндегидрогеназы, ничтожна, поэтому синтез всех остальных протеиногенных аминокислот путем восстановительного аминирования практического значения не имеет. Только аланин и глутаминовая кислота возникают таким способом из пировиноградной и а-кетоглутаровой кислот, являющихся нормальными промежуточными продуктами распада углеводов и жирных кислот.
Следовательно, к первичной аспарагиновой кислоте, синтезируемой путем прямого аминирования, добавляются еще две первичные аминокислоты — аланин и глутаминовая кислота, образующиеся в результате восстановительного аминирования. Остальные аминокислоты образуются в результате реакций переаминирования перечисленных аминокислот с соответствующими кетокислотами, возникающими в процессе обмена веществ, а также путем превращения одних аминокислот в другие (рис. 92). Поэтому аланин, аспарагиновую и глутаминовую кислоты называют первичными аминокислотами, а все остальные — вторичными.
Растительные и животные организмы резко отличаются друг от друга по способности синтезировать аминокислоты. В растениях осуществляется беспрепятственный синтез самых разнообразных аминокислот. Здесь создаются не только все 18 аминокислот, постоянно встречающихся в белках, но и огромное число так называемых «экзотических» аминокислот. Некоторые из них иногда находят в составе белков. В растениях сейчас обнаружено несколько сотен аминокислот, и список их возрастает с каждым годом. Часто та или иная аминокислота присутствует в растениях строго определенного вида и ее наличие может служить надежным таксономическим признаком.
В отличие от этого животные синтезируют далеко не все аминокислоты. Из 18 постоянно встречающихся в белках аминокислот в животном организме синтезируется в среднем только половина их, а остальные — не синтезируются. Первые (синтезируемые) называются заменимыми аминокислотами, вторые (несинтезируемые) — незаменимыми. Между различными видами животных есть некоторые отличия в наборе заменимых и незаменимых аминокислот, но в большинстве случаев к незаменимым аминокислотам относятся валин, лейцин, изолейцин, треонин, метионин, лизин, фенилаланин и триптофан. Любопытно, что у заменимых аминокислот, по подсчетам Ю. А. Жданова (1968), в большинстве случаев степень окисления атомов углерода отрицательна, а у незаменимых — всегда положительна. Это свидетельствует о том, что заменимые аминокислоты эволюционно более молоды (возникли в окислительной атмосфере планеты) по сравнению с незаменимыми.
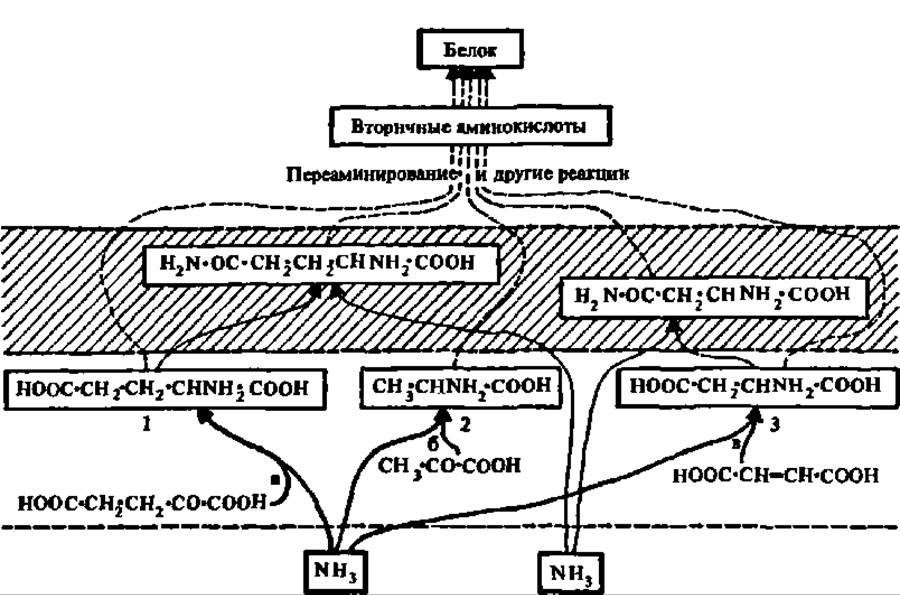
Рис. 92. Взаимосвязь реакций, лежащих в основе биосинтеза первичных и вторичных аминокислот:
1, 2, 3 — первичные аминокислоты: глутаминовая кислота, аланин и аспарагиновая кислота; а — глутамат-дегидрогеназа, 6 — аланиндегидрогеназа, в — аспаргаза
Если в корме животных недостаточно содержание одной или нескольких незаменимых аминокислот, то нормальное развитие животного нарушается, так как биосинтез белка у него идет на низком уровне. Как правило, растительные белки содержат мало лизина, метионина и триптофана. Дефицит этих аминокислот в корме сельскохозяйственных животных встречается наиболее часто. Рационы их неполноценны также по количеству треонина. Введение в рационы недостающих незаменимых аминокислот позволяет нормализовать рост организма, увеличивает привес на каждую израсходованную кормовую единицу, улучшает использование белков основной диеты, резко повышает эффективность животноводства. Так, введение в рацион 0,2—0,5% лизина повышает продуктивность свиноводства и птицеводства на 10—13% и сокращает расход кормового белка на 25%.
Вполне понятно, что в описанных ситуациях речь идет о незаменимых аминокислотах L-ряда, поскольку они необходимы для синтеза белка. Однако L-аминокислоты очень трудно создать путем химического синтеза, при котором получаются рацематы аминокислот, нуждающиеся в разделении на оптические антиподы. Поэтому основная масса аминокислот для нужд животноводства производится путем микробиологического синтеза, т. е. использования определенных микроорганизмов — продуцентов аминокислот, которые выделяют те или иные L-аминокислоты прямо в культуральную жидкость в количестве нескольких граммов на 1 л. В частности, в Институте биохимии им. А. Н. Баха под руководством чл.-кор. В. Н. Букина разработан экономичный микробиологический метод получения L-лизина, а во Всесоюзном научноисследовательском институте генетики и селекции промышленных микроорганизмов его сотрудниками под руководством чл.-кор. В. Г. Дебабова методами генетической инженерии создан штамм кишечной палочки, выделяющий в культуральную среду до 30 г L-треонина. Ряд аминокислот получают также при помощи иммобилизованных бактериальных клеток и ферментов (см. гл. III). Лишь метионин синтезируют заводским путем в виде рацемата, который столь же хорошо используется организмом, как и L-метионин. В нашей стране путем микробиологического синтеза готовят лизин на ряде биохимических заводов с конечной целью довести его производство до 32 тыс. т в год.
Проблема незаменимых аминокислот актуальна и в питании человека, которому необходимо ежедневно получать с пищей 1 г L-триптофана, 2—3 г L-треонина, по 2—4 г L-лейцина, L-метионина и L-фенилаланина, 3—4 г L-изолейцинаи 3—5 г L-лизина. По мнению акад. А. Н. Несмеянова, высказанному в докладе на IX Менделеевском съезде по общей и прикладной химии четверть века тому назад, индустриальный синтез восьми незаменимых аминокислот, способных заменить белок в питании человека, представляется вполне реальным и экономически оправданным. За прошедшие годы промышленное производство как незаменимых, так и заменимых L-аминокислот продвинулось далеко вперед и синтез некоторых из них занимает все более прочное место в ряду мероприятий, направленных на повышение полноценности белкового питания человека.