ОСНОВЫ БИОХИМИИ ЛЕНИНДЖЕРА - ТОМ 1. ОСНОВЫ БИОХИМИИ СТРОЕНИЕ И КАТАЛИЗ - 2011
ЧАСТЬ I. СТРОЕНИЕ И КАТАЛИЗ
12. БИОСИГНАЛИЗАЦИЯ
12.12. Онкогены, гены опухолевых супрессоров и программируемая гибель клетки
Опухоли и рак возникают в результате неконтролируемого клеточного деления. В норме деление клеток регулируется семейством внеклеточных ростовых факторов — белков, которые заставляют покоящиеся клетки делиться и в некоторых случаях дифференцироваться. В результате устанавливается баланс между образованием новых клеток и разрушением старых (клетки кожи отмирают и заменяются новыми каждые несколько недель; лейкоциты заменяются на новые через несколько дней после образования). Если из-за дефектов регуляторных белков баланс «старые клетки — новые клетки» нарушается, может возникнуть клон клеток, деление которых не поддается регуляции (опухоль), а в какой-то момент их присутствие начинает мешать функционированию нормальных тканей (злокачественная опухоль, или рак). Непосредственной причиной возникновения рака почти всегда является генетический дефект одного или нескольких белков, регулирующих деление клеток. Иногда дефектный ген наследуется от одного из родителей; в других случаях мутация происходит в результате действия токсичного вещества (мутагена или канцерогена) или проникающей радиации на ДНК одной-единственной клетки. В большинстве случаев действуют как наследственные факторы, так и факторы окружающей среды; кроме того, в большинстве случаев для полного нарушения регуляции деления клеток и развития рака требуется несколько мутаций. Нарушения в синтезе, регуляции или распознавании факторов роста могут привести к развитию рака.
Онкогены — это мутантные формы генов белков, регулирующих клеточный цикл
■ Онкогены первоначально были открыты в вирусах, вызывающих опухоли. Позже было обнаружено, что они очень близки генам (а может быть, и произошли от них) клеток животных — протоонкогенам, которые кодируют белки, регулирующие рост. В процессе вирусной инфекции последовательность ДНК протоонкогена иногда копируется вирусом и внедряется в его геном. В определенный момент в ходе развития вирусной инфекции ген может стать дефектным в результате укорачивания или мутации. У вирусов, в отличие от животных клеток, нет эффективных механизмов исправления ошибок во время репликации ДНК, поэтому мутации у них быстро накапливаются. Когда вирус, несущий онкоген, заражает клетку хозяина, вирусная ДНК (и онкоген) включается в ДНК клетки хозяина, где теперь может влиять на регуляцию этого деления. По альтернативному невирусному механизму в отдельной клетке ткани, подвергшейся действию канцерогенов, может повредиться ДНК, что приводит к образованию дефекта в одном из регуляторных белков. Далее тот же онкогенный эффект: нарушение регуляции клеточного деления.
Мутации, продуцирующие онкогены, являются генетически доминантными; если любая из пар хромосом содержит дефектный ген, продукт этого гена посылает сигнал деления; результат этого — развитие опухоли. Онкогенный дефект может быть в любом из белков, участвующих в передаче сигнала деления. Известны онкогены, которые кодируют секретируемые белки, факторы роста, трансмембранные белки (рецепторы), цитоплазматические белки (G-белки и протеинкиназы) и ядерные факторы транскрипции, контролирующие экспрессию генов, необходимых для клеточного деления (Jun, Fos).
Дополнение 12.5. МЕДИЦИНА. Создание противоопухолевых лекарственных препаратов на основе ингибиторов протеинкиназ
Если отдельная клетка начинает делиться без какой- либо регуляции и ограничений, это неизбежно приведет к появлению столь значительного клона клеток, что они будут мешать выполнению нормальных физиологических функций организма (рис. 1). Это и есть рак основная причина смертности в развитых странах; и эта причина смертности скоро станет основной и в развивающихся странах. При любом типе рака нормальная регуляция деления клетки нарушается из-за дефекта одного или нескольких генов. Например, гены белков, в норме посылающих периодический сигнал для деления клеток, становятся онкогенами, кодирующими конститутивно активные сигнальные белки. А если мутируют гены белков, останавливающих деление клеток (гены опухолевых супрессоров), образуются белки, не выполняющие этой ограничительной функции. Во многих типах рака обнаруживаются оба варианта мутаций.
Рис. 1. Нарушение регуляции одной-единственной клетки толстой кишки приводит к развитию первичной раковой опухоли с метастазами в печени. При вскрытии выявлены вторичные опухоли, которые выглядят как белые вкрапления в ткани печени.

Многие онкогены и гены опухолевых супрессоров кодируют протеинкиназы или белки, действующие в сигнальных путях перед протеинкиназами. В связи с этим можно надеяться, что специфические ингибиторы протеинкиназ окажутся полезными для лечения опухолевых заболеваний. Например, мутантная форма рецептора EGF представляет собой постоянно активированную рецепторную тирозинкиназу (RTK), передающую сигнал деления клеток вне зависимости от наличия EGF (см. рис. 12-49). Приблизительно у 30% женщин с метастазирующим раком груди в результате мутации гена рецептора HER2/neu образуется RTK с активностью, которая в 100 раз превышает норму. Для образования новых кровеносных сосудов (ангиогенез), чтобы обеспечить солидную опухоль кровью, должна быть активирована еще одна RTK — рецептор фактора роста сосудистого эндотелия (VEGF-R), так что ингибирование VEGF-R может оставить опухоль без необходимых для се развития питательных веществ. Нерецепторные тирозинкиназы также могут мутировать, что приводит к постоянной передаче сигнала и нарушению регуляции клеточного деления. Например, онкоген Аbl (от англ. Abelsonleukemia) связан с развитием острого миелоидного лейкоза — сравнительно редкого заболевания крови (в США около 5000 случаев в год). Онкогены, относящиеся к другой группе, кодируют циклинзависимые протеинкиназы, не поддающиеся регуляции. В каждом из названных выше случаев специфический ингибитор протеинкиназы мог бы оказаться очень ценным лекарственным препаратом для химиотерапевтического лечения заболевания. Поэтому нет ничего удивительного в том, что для создания подобных препаратов прилагаются колоссальные усилия. Однако, как же подойти к решению этой проблемы?
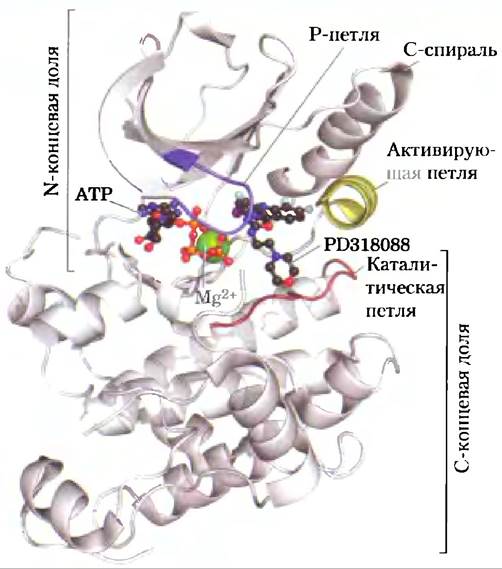
Протеинкиназы всех видов обладают удивительным сходством структуры активного центра. Все имеют такую же структуру, как прототип всех протеинкиназ — РКА (см. рис. 2): две доли, охватывающие активный центр, Р-петля, необходимая для ориентирования и связывания фосфатных групп АТР, активирующая петля, при перемещении которой активный центр становится доступен для белкового субстрата, а также С-спираль, которая меняет свое расположение при активации фермента, приводя аминокислотные остатки субстратсвязывающего кармана в позицию, необходимую для осуществления связывания.
Рис. 2. Основные особенности структуры активного центра протеинкиназ (PDB ID 1S9I). Активный центр фермента окружен N-концевой и С-концевой долями белка; кроме того, в белке содержится каталитическая петля и участок связывания АТР. Активирующая петля этой и других протеинкиназ подвергается фосфорилированию, а затем покидает активный центр, открывая субстратсвязывающий карман, который здесь изображен занятым специфическим ингибитором этого фермента — Р318088. Р-петля необходима для связывания АТР; С-спираль также должна быть расположена правильным образом для связывания АТР и проявления протеинкиназной активности.
Простейшие ингибиторы протеинкиназ это аналоги АТР, которые занимают АТР-связывающий центр, но не могут служить донорами фосфатных групп. Известно множество подобных веществ, однако их клиническое применение ограничено из-за отсутствия селективности: они ингибируют практически любые протеинкиназы, так что оказываемое ими побочное действие не позволяет использовать их в качестве лекарственных средств. Большей селективностью обладают вещества, которые занимают лишь часть АТР-связывающего центра, но за его пределами вступают во взаимодействия с участками белков, уникальными для той протеинкиназы, действие которой нужно заблокировать. Третья возможная стратегия основана на том факте, что, хотя активные конформации всех протеин- киназ одинаковы, их неактивные конформации различаются. Лекарственные препараты, взаимодействующие со специфической протеинкиназой в неактивной конформации и предотвращающие ее переход в активную конформацию, обладают высокой специфичностью. Четвертый подход к решению проблемы основан на использовании специфических антител. Например, моноклональные антитела (с. 254), связывающиеся с внеклеточным участком специфических рецепторных тирозинкиназ, способны уничтожать протеинкиназную активность рецепторов, предотвращая димеризацию или удаляя рецепторы с поверхности клетки. В некоторых случаях антитела, селективно связывающиеся с поверхностью раковых клеток, заставляют иммунную систему атаковать эти клетки.
В период между 1998 г. и серединой 2006 г. в США к применению в противоопухолевой терапии было допущено лишь восемь новых лекарственных препаратов: пять малых молекул и три моноклональных антитела, каждое из которых доказало свою эффективность в клинических испытаниях. Например, иматиниба мезилат (гливек; рис. 3, а) — маленькая молекула с ингибирующими свойствами — практически со 100%-й эффективностью вызывает ремиссию у пациентов с хроническим миелолейкозом, обнаруженным на ранней стадии. Эрлотиниб (тарцева; рис. 3,6), взаимодействующий с EGF-R, оказался эффективен в лечении немелкоклеточного рака легкого на поздних стадиях заболевания. Поскольку во многих системах передачи сигнала, задействованных в делении клеток, принимают участие не одна, а несколько протеинкиназ, для лечения рака могут оказаться полезными те ингибиторы, которые блокируют действие нескольких протеинкиназ. Сунитиниб (сутент) и сорафениб (нексавар) действуют на несколько протеинкиназ, включая VEGF-R и PDGF-R. Эти два препарата применяются для лечения больных с опухолью гастроинтестинальной стромы и прогрессирующим раком почки соответственно. Трастузумаб (герцептин), цетуксимаб (эрбитукс) и бевацизумаб (авастин) — это моноклональные антитела, взаимодействующие соответственно с НER2/neu, EGF-R и VEGF-R; все три препарата применяют для лечения различных типов рака.
Рис. 3. Взаимодействие некоторых ингибиторов протеинкиназ, проходящих клинические испытания или уже используемых в клинической практике, с белками-мишенями. а) Иматиниб связывается в активном центре онкогенной протеинкиназы Abl (PDB ID 1IEP); он занимает как центр связывания АТР, так и прилежащий к нему участок. б) Эрлотиниб связывается в активном центре EGF-R (PDB ID 1М17). в, г) Флавопиридол — ингибитор циклинзависимой протеинкиназы CDK2; показано нормальное связывание АТР (в) в активном центре (PDB ID 1S9I) и связывание флавопиридо- ла (г), препятствующее связыванию АТР (PDB ID 2A4L).
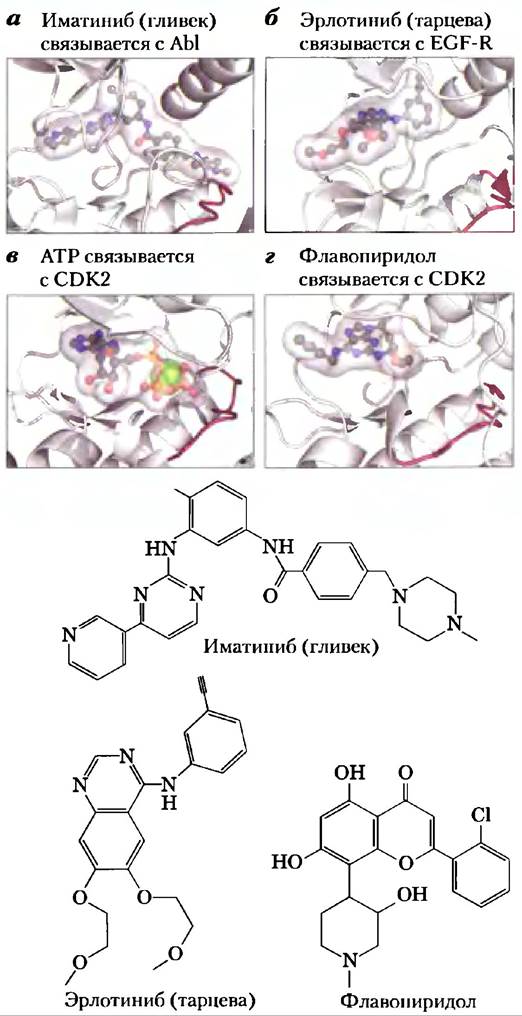
Не менее сотни других соединений находятся сейчас на стадии доклинических испытаний. Среди этих препаратов некоторые получены из природных источников, другие — синтетическим путем. Индирубин входит в состав китайского травяного сбора, традиционно применяемого для лечения некоторых вариантов лейкоза; он ингибирует CDK2 и CDK5. Флавопиридол (рис. 3, г) — синтетический аналог алкалоида, выделяемого из коры индийского растения Amoora rohïtuka, является общим ингибитором всех CDK. Сейчас на клинические испытания направляются несколько сотен потенциально противоопухолевых препаратов, и это вселяет надежду на получение более эффективных и более специфичных препаратов, чем те, которыми мы располагаем в настоящий момент.
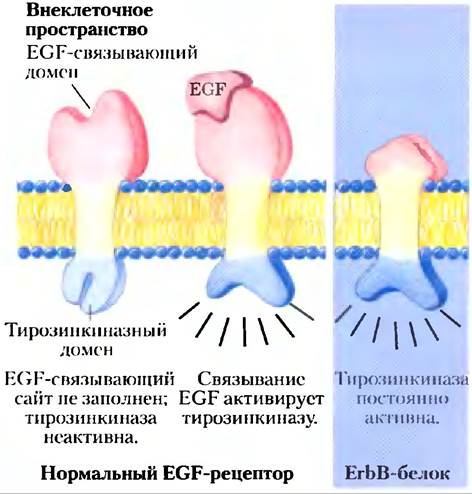
Некоторые онкогены кодируют поверхностные рецепторы с дефектными или отсутствующими участками связывания сигнальных веществ, например, такие, у которых не регулируется тирозинкиназная активность. Так, белок ЕrbВ по существу идентичен нормальному рецептору эпидермального фактора роста, за исключением того, что у ErbB отсутствует N-концевой домен, в норме связывающий EGF (рис. 12-49); в результате он посылает сигнал деления независимо от того, присутствует рецептор EGF или нет. Мутации в еrbВ2, гене для рецепторной Tyr-киназы, сходном с ErbB, обычно связаны со злокачественными опухолями железистого эпителия в молочной железе, желудке и яичниках. (По поводу сокращений, использованных в наименованиях генов и их продуктов, см. гл. 25.)
Рис. 12-49. Кодируемый онкогеном дефектный EGF-рецептор. Продукт онкогена еrbВ (белок ЕrbВ) представляет собой «урезанный вариант» нормального рецептора эпидермального фактора роста (EGF). Его внутриклеточный домен имеет структуру, которая в норме создается связыванием с EGF, но данному белку недостает внеклеточного центра связывания EGF. Без регуляции со стороны EGF белок ЕrbВ непрерывно подает сигнал клеточного деления.
Поскольку протеинкиназы играют ключевую роль в передаче сигнала при нормальном и аномальном делении клетки, именно эти ферменты стали одной из главных мишеней для действия лекарственных препаратов, направленных на лечение опухолевых заболеваний (доп. 12-5). Мутантные формы G-белка Ras широко распространены в опухолевых клетках. Онкоген ras кодирует белок, способный нормально связывать GTP, но не имеющий GТРазной активности. Мутантный белок Ras, таким образом, всегда находится в активированной (связанной с GTP) форме, несмотря на сигналы, поступающие через рецепторы. В результате этого может происходить нерегулируемый рост клеток. Мутациями ras обусловлены от 30% до 50% случаев рака легких и толстой кишки и более чем 90% случаев рака поджелудочной железы. ■
Дефекты в генах опухолевых супрессоров приводят к устранению нормальных ограничителей клеточного деления
Гены опухолевых супрессоров кодируют белки, которые в норме ограничивают клеточное деление. Мутация в одном или нескольких таких генах может приводить к образованию опухоли. Нерегулируемый рост из-за дефектов в генах опухолевых супрессоров, в отличие от нерегулируемого роста, обусловленного онкогенами, имеет рецессивный механизм; опухоли образуются, только если обе парные хромосомы содержат дефектный ген. У людей, которые наследуют одну правильную и одну дефектную копию, каждая клетка имеет одну дефектную копию гена. Если любая из 1012 соматических клеток подвергнется мутации в одной «хорошей»
копии, из этой дважды мутантной клетки может развиться опухоль. В результате мутаций в обеих копиях генов белков рRb, р53 или р21 возникают клетки, в которых отсутствует нормальный ограничитель клеточного деления, в результате чего развивается опухоль.
Ретинобластома возникает у детей и вызывает слепоту, если ее не лечить хирургически. В клетках ретинобластомы два дефектных аллеля гена Rb. Маленькие дети, у которых развивается ретинобластома, обычно имеют множественные опухоли в обоих глазах. Эти дети унаследовали одну дефектную копию гена Rb, которая присутствует в каждой клетке; каждая опухоль происходит из единичной клетки сетчатки, в которой произошла мутация одной «хорошей» копии гена Rb. (Плод с двумя мутантными аллелями в каждой клетке не выживает.) У людей, у которых в детстве была ретинобластома, высока вероятность возникновения злокачественных опухолей легкого, простаты и молочной железы.
Гораздо менее вероятна ситуация, когда у человека, рожденного с двумя «хорошими» копиями гена, произойдут две независимые мутации в обеих копиях гена в одной и той же клетке, тем не менее это случается. У некоторых людей ретинобластома развивается позднее, обычно при этом опухоль бывает только в одном глазу. Эти люди, по-видимому, родились с двумя хорошими копиями (аллелями) Rb в каждой клетке, но оба гена Rb в одной клетке сетчатки подверглись мутации, приводящей к развитию опухоли. У детей после трех лет клетки сетчатки перестают делиться, так что возникновение ретинобластомы в более позднем возрасте — явление достаточно редкое.
Ответственные за стабильность генома «гены-смотрители» кодируют белки, которые участвуют в репарации большинства генетических дефектов, возникающих из-за нарушений репликации ДНК, действия ионизирующей радиации или канцерогенов из окружающей среды. Мутации данных генов приводят к повышенной частоте нерепарируемых повреждений (мутаций) других генов, включая протоонкогены и гены опухолевых супрессоров, что, в свою очередь, приводит к развитию рака. К «генам-смотрителям» относятся ген АТМ (см. рис. 12-48), семейство генов ХР, мутации которых приводит к развитию пигментной ксеродермы, а также гены ВRСА1, свя-
занные с некоторыми типа рака молочной железы (см. доп. 25-1). Мутации в гене белка р53 также вызывают опухоли. Более чем в 90% случаев плоскоклеточного рака кожи и в 50% всех остальных видов рака у человека ген р53 дефектный. Те редкие люди, которые наследуют одну дефектную копию р53, обычно страдают раковым синдромом Ли-Фраумени, при котором с высокой частотой и в раннем возрасте возникают множественные злокачественные опухоли (молочной железы, мозга, костей, крови, легких и кожи). Объяснение возникновения множественных опухолей в этих случаях то же самое, что и при мутациях гена Rb: у человека, рожденного с одной дефектной копией р53 в каждой соматической клетке, вероятно, в течение жизни будет появляться вторичная мутация р53 более чем в одной клетке.
Таким образом, развитие рака может быть связано с тремя группами причин: дефекты онкогенов можно сравнить с постоянно нажатой педалью акселератора автомобиля при работающем моторе, мутации генов опухолевых супрессоров можно сравнить с неработающими тормозами, а мутации «генов-смотрителей», приводящие к отсутствию репарации механизма репликации, сравнимы с неквалифицированным механиком, обслуживающим ваш автомобиль.
Мутации в онкогенах и генах опухолевых супрессоров не обладают эффектом «все или ничего». При некоторых опухолях, а возможно, при всех, переход от нормальной клетки до злокачественной опухоли требует накопления мутаций (иногда в течение нескольких десятилетий), ни одна из мутаций в отдельности не несет ответственности за конечный эффект. Например, развитие рака прямой кишки имеет несколько распознаваемых стадий, и каждая из них связана с мутацией (рис. 12-50).
Рис. 12-50. От нормальной эпителиальной клетки к раку кишечника. В толстой кишке мутации в обеих копиях гена опухолевого супрессора АРС приводят к доброкачественным новообразованиям — скоплениям эпителиальных клеток, которые слишком быстро размножаются (ранняя аденома). Если клетка, уже имеющая дефект в АРС, подвергается второй мутации в протоонкогене ras, дважды мутантная клетка дает начало промежуточной аденоме, образуя доброкачественный кишечный полип. Когда одна из таких клеток подвергается дальнейшим мутациям в генах опухолевых супрессоров DCC (окончательно не установлено) и р53, образуются опухоли, агрессивность которых все более нарастает. Затем мутации в еще не охарактеризованных генах приводят к злокачественной опухоли и в конечном итоге к метастазирующему раку, который может распространяться в другие ткани. По-видимому, большинство злокачественных опухолей возникают в результате серии мутаций такого рода.
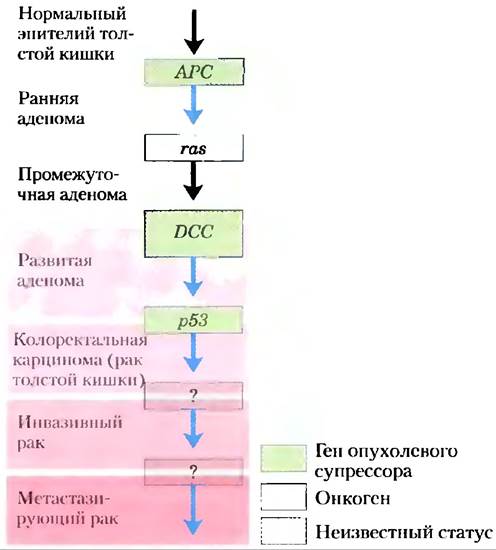
Если нормальная эпителиальная клетка кишечника претерпевает мутации в обеих копиях гена опухолевого супрессора АРС (от англ. adenomatous poliposis coli — аденоматозный полипоз толстой кишки), она начинает делиться быстрее, чем нормальная и производит собственный клон — доброкачественный полип (ранняя аденома). По неизвестным пока причинам мутация АРС приводит к хромосомной нестабильности; целые области хромосомы утрачиваются или перестраиваются в процессе деления клетки. Эта нестабильность может приводить ко второй мутации, обычно в гене ras, которая превращает клон в промежуточную аденому. Третья мутация (вероятно, в гене опухолевого супрессора DCC) приводит к поздней аденоме. Только когда обе копии р53 становятся дефектными, клеточная масса действительно становится карциномой — злокачественной опухолью, опасной для жизни. Весь процесс в целом требует, таким образом, наличия по крайней мере семи генетических нарушений: двух в каждом из трех генов опухолевых супрессоров (АРС, DCC и р53) и одного в протоонкогене ras. Наверное, существуют еще несколько других путей развития колоректального рака, но механизм, по которому злокачественный процесс является результатом множественных мутаций, вероятно, всегда выдерживается. Если полип обнаруживают на стадии ранней аденомы и хирургическим путем удаляют клетки, несущие первые мутации, поздние аденомы и карциномы не разовьются; отсюда очевидно значение ранней диагностики. У клеток и многоклеточных организмов есть система ранней детекции дефектов. Например, белки АТМ и ATR, описанные в разд. 12.11, обнаруживают протяженные повреждения ДНК, которые не могут быть эффективно восстановлены. Далее они запускают процесс апоптоза (с участием р53), в результате которого опасные для организма клетки уничтожают сами себя. ■
Апоптоз — программируемая гибель клетки
Многие клетки могут точно контролировать время своей собственной смерти с помощью процесса программируемой клеточной гибели или апоптоза (от греч. apoptosis — опадание листьев). Например, при развитии зародыша некоторые клетки должны отмереть. Формирование пальцев из коротких зачатков конечностей требует хронометрированной гибели клеток между развивающимися костями пальцев. В процессе развития нематоды Caemohabditis elegans из оплодотворенного яйца точно 131 клетка (из общего числа 1090 соматических клеток в эмбрионе) должна подвергнуться запрограммированной смерти для того, чтобы образовался взрослый организм.
Апоптоз имеет значение и в других процессах, а не только в развитии. Когда производящая антитело клетка начинает образование антител против антигена, присутствующего в организме в норме, эта клетка подвергается запрограммированной гибели в тимусе (вилочковой железе). Апоптоз представляет собой необходимый механизм для элиминации аутоантител. Ежемесячное отторжение клеток стенки матки (менструация) представляет собой другой случай гибели нормальных клеток, происходящей путем апоптоза. Иногда самоубийство клетки не запрограммировано, а происходит в ответ на биологические обстоятельства, которые пред
вещают гибель организма. Например, инфицированная вирусом клетка, умирающая до окончания цикла инфицирования, предотвращает распространение вируса на соседние клетки. Разные стрессовые воздействия, такие как высокая температура, гиперосмоляльность, УФ- и гамма-излучения, также приводят в действие механизм клеточного самоубийства; организму в целом лучше, когда поврежденные клетки погибают.
Регуляторные механизмы, запускающие апоптоз, основаны на действии некоторых пептидов, которые регулируют клеточный цикл. Сигнал к самоубийству часто приходит извне, через поверхностный рецептор. Фактор некроза опухолей (TNF, от англ. tumor necrosis factor), продуцируемый клетками иммунной системы, взаимодействует с клетками посредством специфических TNF-рецепторов. Эти рецепторы имеют TNF-связывающие центры на внешней поверхности плазматической мембраны и «домен смерти», содержащий около 80 аминокислотных остатков, который передаст сигнал к самоликвидации через мембрану на цитозольные белки, такие как TRADD (от англ. TNF receptor-associated death domain; рис. 12-51). Другой рецептор. Fas, имеет похожий домен смерти, позволяющий ему взаимодействовать с цитозольным белком FADD (от англ. Fas-associated death domain), который активирует цитозольную протеазу, называемую каспазой 8. Этот белок принадлежит к семейству протеаз, участвующих в апоптозе; все они синтезируются в виде неактивных проферментов, все содержат ключевой остаток Cys в активном центре и все гидролизуют белки-мишени по С-концу специфического остатка Asp (отсюда и название «каспазы», от Cys и Asp).
Рис. 12-51. Основные события апоптоза. Рецепторы плазматической мембраны (Fas, TNF-R1) получают сигналы извне (от лиганда Fas или фактора некроза опухолей TNF соответственно). Активированный рецептор обеспечивает взаимодействие между «доменом смерти» (последовательность из 80 аминокислот) в Fas или TNF-R1 и похожим доменом смерти в цитозольных белках FADD или TRADD. FADD активирует цитозольную протеазу каспазу 8, которая протеолитически активирует другие клеточные протеазы. TRADD также активирует протеазы. Итоговый протеолиз является главным фактором гибели клетки.
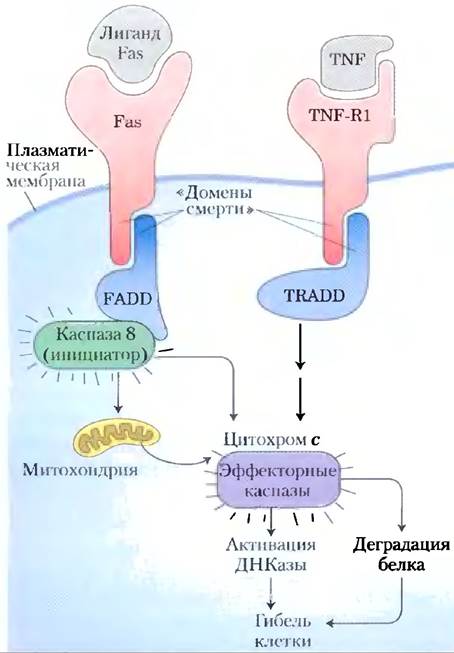
Каспаза 8, каспаза-«ипициатор», активируемая апоптотическим сигналом, передаваемым через FADD, затем самоактивируется, расщепляя собственный профермент. Мишенями активной каспазы 8 могут быть митохондрии. Протеаза приводит к высвобождению некоторых белков, содержащихся между наружной и внутренней митохондриальными мембранами: цитохрома с (гл. 19) и нескольких эффекторных каспаз. Цитохром с связывается с проферментной формой эффекторной каспазы 9 и стимулирует ее протеолитическую активацию. Активированная каспаза 9 в свою очередь катализирует деструкцию клеточных белков, а это и является главной причиной гибели клеток в результате апоптоза. Одной из специфических мишеней действия каспазы является активируемая каспазой дезоксирибонуклеаза.
При апоптозе мономерные продукты деградации белка и ДНК (аминокислоты и нуклеотиды) высвобождаются в результате контролируемого процесса, который делает возможным их улавливание и использование соседними клетками. Таким образом, апоптоз позволяет организму удалить клетку без потери ее компонентов.
Краткое содержание раздела 12.12 Онкогены, гены опухолевых супрессоров и программируемая гибель клетки
■ Онкогены кодируют дефектные сигнальные белки. Непрерывно подавая сигнал к делению клеток, они приводят к образованию опухоли. Онкогены являются генетически доминантными и могут кодировать дефектные факторы роста, рецепторы, G-белки, протеинкиназы или ядерные регуляторы транскрипции.
■ Гены опухолевых супрессоров кодируют регуляторные белки, которые в норме тормозят клеточное деление; мутации в этих генах генетически рецессивны, но могут приводить к образованию опухоли.
■ Рак возникает обычно как результат накопления мутаций в онкогенах и генах опухолевых супрессоров.
■ При мутации «генов-смотрителей», кодирующих белки, необходимые для репарации генетических повреждений, остаются без репарации повреждения в других генах, включая мутации протоонкогенов и генов опухолевых супрессоров, что может привести к развитию рака.
■ Апоптоз может запускаться внеклеточными сигнальными веществами, такими как TNF, через рецепторы плазматической мембраны.