Биохимия аминокислот - А. Майстер 1961
Природные аминокислоты
Аминокислоты, обычно встречающиеся в белках
Введение
«При более тщательном исследовании продуктов растительного происхождения современные химики обнаружили множество соединений, не известных более ранним исследователям; однако давно уже, я полагаю, не находили в растениях соединения столь исключительного и интересного, как то, на котором мы теперь остановимся... В некотором количестве сока спаржи, сконцентрированного выпариванием, я обнаружил довольно большое число кристаллов, два вида которых, как мне кажется, принадлежат новым веществам; так как эти кристаллы различались по форме, по прозрачности и по вкусу, для меня не представило трудностей разделить их». Вокелен (1806).
Известны 22 аминокислоты, которые регулярно или часто встречаются в гидролизатах белков. Открытие этих аминокислот явилось важным этапом в развитии современной биохимии. Более детальные сведения по истории интереснейших капитальных работ, приведших к обнаружению этих аминокислот, изложены в обзоре Виккери и Шмидта [1]. По мнению названных авторов, наличие той или иной аминокислоты в белковых гидролизатах можно считать установленным лишь в том случае, если она была выделена по крайней мере двумя исследователями независимо друг от друга и если ее строение подтверждено синтезом. Эти критерии и в настоящее время сохраняют свое значение при оценке сообщений о вновь открываемых аминокислотах. Между тем их нельзя считать непогрешимыми; об этом свидетельствует хотя бы тот факт, что Виккери и Шмидт сами впали в ошибку, отнеся ß-оксиглутаминовую кислоту к числу «признанных» продуктов гидролиза белков (стр. 88). К категории «признанных» аминокислот в течение известного времени причисляли также и норлейцин.
Аминокислоты, рассматриваемые ниже, многократно выделялись из белковых гидролизатов, и их структура окончательно установлена. В этот список вошли все аминокислоты, перечисленные в 1931 г. Виккери и Шмидтом, за исключением β-окси- глутаминовой кислоты и йодсодержащих аминокислот, и, кроме того, аспарагин, глутамин, цистеин и треонин. Следует отметить, что использованное в тексте выражение ««обычно обнаруживаемые« в гидролизатах аминокислоты» до известной степени произвольно и допускает некоторое расхождение в толковании. Так, например, известно, что 3,5-дийодтирозин и тироксин присутствуют в тиреоглобулине; точно установлено наличие в некоторых белках 8-оксилизина. Эти аминокислоты и некоторые другие, реже встречающиеся в белковых гидролизатах, будут рассмотрены в соответствующем разделе (стр. 62).
Аминокислоты, обсуждаемые в настоящем разделе, приведены в алфавитном порядке. Они могут быть классифицированы следующим образом:
Алифатические аминокислоты
Моноаминомонокарбоновые
Глицин
Аланин
Изолейцин
Лейцин
Валин
Оксимоноаминомонокарбоновые
Серин
Треонин
Моноаминодикарбоновые
Аспарагиновая кислота
Глутаминовая кислота
Амиды моноаминодикарбоновых кислот
Аспарагин
Глутамин
Диаминомонокарбоновые
Аргинин
Лизин
Серусодержащие
Цистеин и цистин
Метионин
Ароматические аминокислоты
Фенилаланин
Тирозин
Гетероциклические аминокислоты
Триптофан
Гистидин
Пролин
Оксипролин
L-Аланин (а-аминопропионовая кислота)
![]()
Аланин принадлежит к числу тех аминокислот, которые сначала были получены синтетически и лишь позднее признаны природными продуктами. В 1850 г. Штреккер [2], пытаясь получить молочную кислоту, обработал продукт конденсации ацетальдегида и аммиака цианистоводородной и соляной кислотами; полученный в кристаллической форме аланин был превращен в молочную кислоту путем обработки азотистой кислотой. Реакция Штреккера ведет к образованию аминонитрила, который после гидролиза дает соответствующую аминокислоту; оказалось, что эта реакция может быть использована для получения ряда других аминокислот из соответствующих альдегидов. Через 38 лет после того, как Штреккер синтезировал аланин, Вейл [3] выделил эту аминокислоту из кислотного гидролизата шелка — белка, наиболее богатого аланином. Позднее Фишер и Скита [4] получили L-аланин из шелка и установили его структуру и конфигурацию путем превращения его в молочную кислоту.
L-Аргинин (а-амино-δ-гуанидиновалерьяновая кислота)
![]()
Шульце и Штайгер [5] выделили аргинин из этиолированных проростков люпина в 1866 г. В 1895 г. Хедин [6] сообщил о получении азотносеребряной соли аргинина из гидролизатов рога. Впоследствии Коссель и Гросс [7] нашли, что аргинин является одной из главных составных частей основных белков спермы рыб. Строение аргинина было установлено путем его щелочного гидролиза на орнитин и мочевину [8] и синтезом из бензоилорнитина [9].
Во время кислотного гидролиза белков аргинин может расщепляться до орнитина, благодаря чему последний иногда обнаруживается в белковых гидролизатах. Описано также превращение аргинина в цитруллин при щелочном гидролизе [10]. При обработке 1-нафтолом и гипохлоритом или гипобромитом натрия аргинин дает красную окраску (реакция, впервые описанная Сакагути [11]). Аргинин может быть осажден из растворов в виде моно- или дифлавианата [7]; этой реакцией пользуются для выделения аргинина из белковых гидролизатов.
Аргинин не только содержится в белках, но встречается и в свободном состоянии, а также в виде фосфоаргинина
![]()
в мышцах беспозвоночных, где это соединение выполняет функцию, аналогичную функции фосфокреатина у высших животных [12, 13]. Кроме того, аргинин входит в состав октопина (стр. 57) и аргининоянтарной кислоты (стр. 339). Родственное ему в химическом отношении соединение, канаванин, было выделено из бобов Canavalia (стр. 49)
L-Аспарагин (β-амид а-аминоянтарной кислоты)
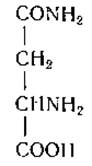
Аспарагин был первой аминокислотой, выделенной из природных продуктов. В 1806 г. Вокелен и Робике [14] получили его из сока спаржи. Кислотный гидролиз белка приводит к дезамидированию аспарагина в аспарагиновую кислоту. Наличие аммиака в кислых гидролизатах белка привело Глазивеца и Габермана в 1873 г. [15] к предположению, что источником этого аммиака могут служить амидные группы глутамина и аспарагина. Однако наличие аспарагина в белках было доказано лишь в 1932 г., когда Дамодаран [16] описал выделение аспарагина из ферментных гидролизатов эдестина.
Аспарагин представляет собой широко распространенное соединение; он накапливается в значительных концентрациях у некоторых видов высших растений, а также встречается в свободном состоянии в тканях животных. При расщеплении белков под действием кислот происходит гидролиз аспарагина; однако его амидная группа относительно более устойчива, чем амидная группа глутамина.
В отличие от большинства аминокислот, реагирующих с нингидрином с образованием продукта; окрашенного в пурпурный цвет, аспарагин и некоторые другие ß-аспартилпроизводные дают при реакции с нингидрином коричневую окраску. Было отмечено, что появление коричневой окраски при реакции нингидрина с аспарагином не сопровождается выделением СО2 [17]. Данные, полученные при изучении инфракрасных спектров [18] и дифракции рентгеновских лучей [19, 20], свидетельствуют о наличии внутримолекулярного взаимодействия между амидной и карбоксильной группами молекулы аспарагина; не исключено, что этим взаимодействием обусловлено аномальное поведение аспарагина при реакции с нингидрином.
Предположение Стюарда и Томпсона [17], что аспарагин может существовать в виде гидрата циклического имида, не подтвердилось, поскольку было установлено, что синтетический имид а-аминоянтарной кислоты не идентичен аспарагину [21].
L-Аспарагиновая кислота (а-аминоянтарная кислота)
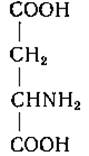
Аспарагиновая кислота была сначала описана как продукт гидролиза аспарагина. Позднее Ритгаузен [22] выделил аспарагиновую кислоту из белкового гидролизата. Пириа [23] получил яблочную кислоту из аспарагиновой кислоты воздействием на последнюю азотистой кислотой, а затем был осуществлен и синтез аспарагиновой кислоты [24, 25].
N-Ацетил-L-аспарагиновая кислота найдена в экстрактах мозга кошки в концентрациях порядка 100 мг на 100 г ткани; она содержится также в мозге крысы и в меньших количествах (от 1 до 3 мг на 100 г) в печени, почках, мышцах и моче кошки [26].
L-Валин (a-аминоизовалерьяновая кислота)
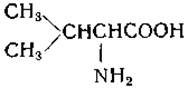
Валин был открыт в экстрактах поджелудочной железы Горуп-Безанессом в 1856 г., [95], однако первым, кто показал, что валин является продуктом гидролиза белка (альбумина), был Шютценбергер [96]. Строение валина было окончательно выяснено в 1906 г. Фишером [97], который идентифицировал природный валин с одним из стереоизомеров, полученных при разделении синтетической аминокислоты. Валин присутствует во многих белках, но обычно — в относительно малых количествах
L-Гистидин (a-амино-β-имидазолпропионовая кислота)
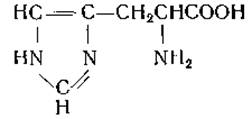
Гистидин был выделен Косселем [50] в 1896 г. из сернокислых гидролизатов стурина (протамин спермы осетра). В том же году Хедин [51] независимо от Косселя выделил гистидин из белковых гидролизатов. Паули доказал наличие имидазольного кольца в молекуле гистидина и нашел, что при взаимодействии гистидина с диазотированной сульфаниловой кислотой в щелочном растворе развивается красное окрашивание (реакция Паули).
В результате работ Паули [52] и других авторов [53, 54] было выяснено строение молекулы гистидина, которое было окончательно доказано синтезом гистидина, осуществленным Пайменом [55] в 1911 г. Гистидин присутствует в относительно больших количествах в гемоглобине; кроме того, он входит в состав эрготионина (в виде тиолгистидина), карнозина и ансерина (стр. 55 и 70).
Глицин (аминоуксусная кислота)
![]()
Глицин явился первой аминокислотой, выделенной из белкового гидролизата. В 1820 г. Браконно [46] получил глицин из сернокислого гидролизата желатины и обратил внимание на сладкий вкус этой аминокислоты. В дальнейшем описанный Браконно «сахар желатины» был назван гликоколлом, а затем глицином. Браконно не знал о наличии азота в молекуле глицина; более поздние работы, завершением которых явились исследования Каура [47, 48], привели к установлению строения глицина и синтезу его из монохлоруксусной кислоты и аммиака.
Глицин присутствует в больших количествах в желатине и входит в состав многих других белков. В виде амида он встречается в окситоцине и вазопрессине (стр. 72). Глицин является составной частью целого ряда природных веществ, например глутатиона, а также гиппуровой и гликохолевой кислот. Помимо этого, в природе встречается N-метилпроизводное глицина, саркозин; было показано, что это вещество является продуктом тканевого обмена у млекопитающих (стр. 329). Саркозин обнаружен также в составе белка земляного ореха [49] и в гидролизатах некоторых антибиотиков (стр. 77).
L-Глутамин (y-амид a-аминоглутаровой кислоты)
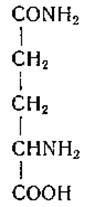
Интересно, что, тогда как выделение аспарагина предшествовало выделению аспарагиновой кислоты, глутамин был впервые получен лишь через 17 лет после обнаружения глутаминовой кислоты в белковых гидролизатах.
Шульце и Босхард [39] получили его из сока свеклы в 1883 г. Глазивец и Габерман [15] впервые высказали предположение о присутствии глутамина в белке; это предсказание относительно недавно подтвердили Дамодаран и др. [40], выделившие глутамин из ферментных гидролизатов эдестина. Первый синтез глутамина осуществили Бергман к др. [41]. Глутамин накапливается в значительных количествах у некоторых видов высших растений и служит одним из главных аминокислотных компонентов крови млекопитающих.
При обработке смесью азотистой и уксусной кислот 1 моль глутамина в отличие от аспарагина и некоторых других амидов дает почти 2 моля азота. Отличительным свойством глутамина является относительно высокая лабильность его амидной группы, в связи с чем он легко подвергается циклизации с образованием аммонийной соли пирролидонкарбоновой кислоты. У пептидов глутамина, в которых а-аминогруппы замещены, амидные группы относительно устойчивы [42—44]. Другие у-глутамилпроизводные (у-глутамилпептиды, у-этиловый эфир глутаминовой кислоты) и гомоглутамин также проявляют склонность к циклизации [45]. Процесс циклизации глутамина и родственных ему соединений катализируется фосфатами и некоторыми другими анионами (стр. 315).
L-Глутаминовая кислота (а-аминоглутаровая кислота)
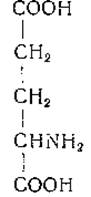
Глутаминовая кислота была выделена Ритгаузеном [35] в 1866 г. из гидролизатов клейковины эндосперма пшеницы. В дальнейшем было показано, что из глутаминовой кислоты при действии азотистой кислоты и последующем восстановлении образуется глутаровая кислота [36, 37]. В 1890 г. Вольф [38] осуществил первый химический синтез глутаминовой кислоты. Эта кислота относится к числу наиболее широко распространенных аминокислот и имеет большое значение в обмене веществ.
Глутаминовая кислота кристаллизуется из водных растворов в присутствии соляной кислоты в виде труднорастворимого хлоргидрата. При кипячении водного раствора этой кислоты она переходит в пирролидон-а-карбоновую кислоту (пироглутаминовая кислота, 5-оксо-2-пирролидинкарбоновая кислота):
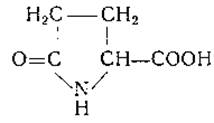
L-Глутаминовая кислота в виде мононатриевой соли находит широкое применение как вкусовая приправа.
L-Изолейцин (а-амино-β-метилвалерьяновая кислота)
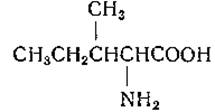
Изолейцин был выделен Эрлихом из свеклосахарной мелассы в 1904 г. Позднее Эрлих выделил эту аминокислоту из неполного гидролизата фибрина, полученного обработкой соком поджелудочной железы, а также из клейковины пшеницы, яичного альбумина и говяжьего мяса.
По наблюдениям Эрлиха, выделенный им продукт имел тот же химический состав, что и лейцин, но по ряду свойств (растворимость, точка плавления, растворимость медной соли) отличался от лейцина. Эрлиху [60—62] удалось расщепить L-изолейцин до d-амиламина и синтезировать эпимер изолейцина из d-изовалеральдегида.
Интересно отметить, что за несколько лет до работ Эрлиха Фишер [63] получил из препаратов «лейцина» фракции, обладающие различной оптической активностью и различной растворимостью.
L-Лейцин (а-аминоизокапроновая кислота)
![]()
Пруст [64] получил лейцин в неочищенной форме из сыра в 1819 г. В 1820 г. Браконно [65] выделил кристаллическую аминокислоту из кислотных гидролизатов мышц и шерсти и назвал ее лейцином. Лейцин был синтезирован посредством реакции Штреккера из изовалеральдегида, и продукт синтеза оказался идентичным рацемизированному природному соединению [66].
L-Лизин (а, ε-диаминокапроновая кислота)
![]()
Лизин был впepвые выделен из гидролизата казеина Дрекселем [67] в 1889 г. Дрексель предполагал, что лизин представляет собой диамин; правильная структура была установлена в 1902 г. Фишером и Вайгертом [68], которые синтезировали лизин и показали, что синтезированный продукт идентичен рацемизированному природному материалу. Содержание лизина в белках варьирует в широких пределах; он часто входит в состав животных белков, но может отсутствовать или содержаться в очень малых количествах в белках растительного происхождения (например, в зеине и глиадине). При обработке белков азотистой кислотой свободные s-аминогруппы лизина превращаются в гидроксильные группы; по-видимому, у лизина, связанного в белках, большинство ε-аминогрупп, если не все, находятся в свободном состоянии (см., однако, стр. 277). Лизин, связанный по ε-NH2-гpyппe, содержится в биоцитине.
L-Метионин (а-амино-у-метилтиомасляная кислота)
![]()
Мюллер [69] открыл эту аминокислоту в 1922 г., пытаясь выяснить природу факторов роста гемолитического стрептококка. Метионин выделили из кислотного гидролизата казеина и определили его элементарный состав. В 1928 г. Барджер и Койн [70] провели синтез метионина при помощи реакции Штреккера и установили строение этого соединения. Впоследствии Виндас и Марвел [71] разделили метионин на оптические антиподы. Интересно, что Осборн [72] задолго до этого отметил наличие в белках двух типов серы — щелочно-лабильной и устойчивой по отношению к щелочам; сера первого типа входит в состав цистеина и цистина, сера второго типа, как теперь известно, принадлежит метионину.
Присутствие метионинсульфоксида в природных объектах обусловлено, вероятно, неферментативным окислением метионина; процесс ферментативного восстановления метионинсульфоксида в метионин описан на стр. 373.
4-Окси-L-пролин (4-оксипирролидин-2-карбоновая кислота)
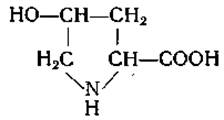
Оксипролин был выделен в 1902 г. из кислых гидролизатов желатины Фишером [56], который путем восстановления гидроксильной группы превратил эту аминокислоту в пролин. Лейксу и его сотрудникам [57—59] удалось синтезировать оксипролин и получить четыре его стереоизомера.
Окси-L-пролин найден только в гидролизатах эластина и коллагена, где на его долю приходится до 13% всех аминокислотных остатков. При обработке нингидрином оксипролин дает на хроматограммах желтую окраску, при обработке изатином он может быть обнаружен в виде синего пятна. Взаимодействие оксипролина с перекисью водорода с последующим подкислением раствора приводит к образованию пиррол-2-карбоновой кислоты (стр. 352), которая дает с n-диметиламинобензальдегидом (реактив Эрлиха) интенсивное красно-фиолетовое окрашивание.
L-Пролин пирролидин-2-карбоновая кислота)
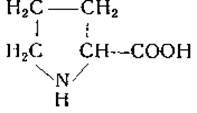
Пролин был синтезирован в 1900 г. Вильштеттєром [75] из эфира а, 5-дибромпропилмалоновой кислоты. В 1901 г. Фишер [76] получил L-пролин и DL-пролин из гидролизатов казеина и показал, что второй продукт идентичен синтетическому пролину, который он приготовил из фталимидопропилмалонового эфира. Пролин содержится в коллагене, желатине и других белках. Интересным его свойством является растворимость в спирте. На бумажных хроматограммах пролин дает при обработке нингидрином желтую окраску, а при действии изатина — синюю.
L-Серин (а-амино-2-оксипропионовая кислота)
![]()
Серин был впервые выделен Кремером в 1865 г. из белка шелка [77]. Кремер отметил, что по своему строению серин близок аланину и цистину, и пришел к выводу, что серин представляет собой оксиаминокислоту. Строение серина было установлено в 1902 г. Фишером и Лейксом, осуществившими его синтез [78]. Серин широко распространен в белках; относительно большое количество этой аминокислоты находится в фиброине шелка. Серин встречается также в виде фосфорного эфира [79—81]:
![]()
Фосфосерин был выделен Липманом из кислотного гидролизата казеина [79]. Положение фосфатной группы в нативном казеине еще окончательно не установлено. Высказано предположение, что в этом белке имеются N-фосфатные связи и что О-фосфатные связи образуются в результате миграции фосфатного остатка при гидролизе белка [82].
L-Тирозин [a-амино-β-(n-оксифенил)пропионовая кислота]
![]()
Тирозин был впервые получен в 1846 г. Либихом [91] при расщеплении казеина щелочью. Позднее тирозин выделили де-Ла-Рю [92] и Бoпп [93], первый — из кошенильной тли, второй — из белков (альбумина, казеина, фибрина). Строение тирозина установили в 1883 г. Эрленмейер и Липп [94] путем его синтеза. Тирозин чрезвычайно трудно растворим в воде, и этим свойством удобно пользоваться для выделения этой аминокислоты из белковых гидролизатов. В составе фибриногена и в моче человека обнаружен тирозин-О-сульфат (стр. 0357).
L-Треонин (а-амино-β-оксимасляная кислота)
![]()
Треонин был получен из кислотных гидролизатов фибрина в 1935 г. Роузом и др. [83]. Работа этих исследователей была направлена на выделение присутствующего в белковом гидролизате фактора, необходимого для роста крыс. Открытие треонина позволило впервые показать, что крысы могут расти на диете, содержащей очищенные аминокислоты. При химическом восстановлении треонина Роуз и его сотрудники получили L-a-аминомасляную кислоту, а путем окисления перевели треонин в D-молочную кислоту. Треонин был синтезирован Картером [84], а затем Вест и Картер [85] получили четыре стереоизомера этой аминокислоты. Как и серин, треонин встречается в форме своего фосфорного эфира [86]. Серин и треонин реагируют с йодной кислотой с образованием глиоксиловой кислоты, аммиака и муравьиного или, соответственно, уксусного альдегида [87]:
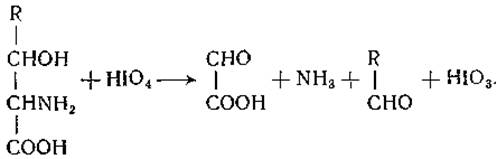
Этой реакцией пользуются для количественного определения упомянутых аминокислот.
L-Триптофан (а-амино-β-3-индолпропионовая кислота)
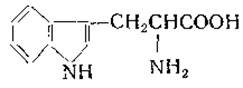
Триптофан был выделен в 1901 г. Гопкинсом и Колом из продуктов переваривания казеина соком поджелудочной железы [88]. До этого Адамкевич [89] наблюдал, что при действии серной кислоты на смесь ледяной уксусной кислоты и альбумина появляется фиолетовая окраска. Гопкинс и Кол нашли, что развитие этой окраски обусловлено наличием глиоксиловой кислоты в препаратах ледяной уксусной кислоты. Авторы попытались выделить из белковых гидролизатов вещество, дающее эту окраску, и в конечном итоге получили триптофан. Строение триптофана было установлено в 1907 г. Эллингером и Фламандом [90]. Эта аминокислота содержится во многих белках, но обычно в небольшом количестве.
Потребность животных в пищевом триптофане по сравнению е потребностью в других аминокислотах относительно невелика (см. стр. 124).
L-Фенилаланин (а-амино-β-фенилпропионовая кислота)
![]()
Фенилаланин был выделен Шульце и Барбьери [73] в 1879 г. из этиолированных побегов люпина. В дальнейшем те же авторы получили эту аминокислоту из гидролизатов растительных белков. В результате химического синтеза фенилаланина, осуществленного в 1882 г. Эрленмейером и Диплом [74], Шульце и Барбьери смогли установить идентичность выделенной ими аминокислоты с синтетическим продуктом [73].
L-Цистеин (а-амино-β-меркаптопропионовая кислота) и L-цистин [β, β',-дитиоди(а-аминопропионовая кислота)]
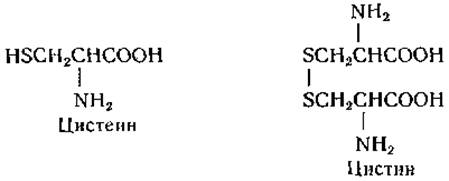
Цистин был выделен Волластоном [27] в 1810 г. из мочевых камней.
В 1899 г. Мёрнер [28] получил цистин из гидролизатов рога. Цистин в значительных количествах входит в состав кератинов и присутствует также во многих других белках. Существуют убедительные доказательства наличия цистеина в составе белков, но в кислотных гидролизатах белков обычно находят только цистин — продукт окисления цистеина. При наличии в белке большого количества триптофана возможно образование цистеина во время гидролиза кислотой [29]; вместе с тем как цистин, так и цистеин разрушаются при обработке щелочью.
О наличии цистеина в некоторых белках свидетельствует тот факт, что эти белки дают положительную цветную (красную) реакцию с нитропруссидом. В нейтральном или щелочном растворе, особенно в присутствии ионов металлов, цистеин быстро окисляется в цистин. Бауман [30] в 1884 г. описал восстановление цистина в цистеин при обработке оловом и соляной кислотой. Эрленмейер [31, 32] установил строение цистина и цистеина путем синтеза этих аминокислот.
Цистеин реагирует с формальдегидом с образованием тиазолидинкарбоновой кислоты:
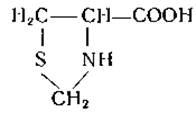
Эта реакция, не свойственная цистину, была использована для раздельного определения цистина и цистеина [33].
Высокоспецифической цветной реакцией на цистеин служит реакция Салливана: цистеин дает характерную красно-коричневую окраску с 1,2-нафтохинон-4-сульфонатом натрия в сильно восстанавливающей среде [34].