Биохимия - Химические реакции в живой клетке Том 1 - Д. Мецлер 1980
Молекулы, из которых мы состоим
Как мы изучаем структуру молекул
Разделение
Каким образом химики установили те тысячи структурных формул, которые мы используем для описания встречающихся в природе молекул? Этот вопрос слишком сложен, чтобы обсуждать его здесь подробно. Но поскольку разделение соединений, анализ смесей и установление структуры молекул по-прежнему занимают важное место в современной биохимии, далее мы даем «мини-обзор» и сопровождаем его рядом ссылок, чтобы помочь читателю ориентироваться в литературе по этим вопросам.
Прежде чем начинать работу над выяснением структуры того или иного вещества, необходимо выделить его в чистом виде из тех сложных смесей, в составе которых оно находится в клетках и тканях, причем зачастую концентрация этого вещества в ткани бывает весьма низкой. Затем молекулы соединения необходимо разрушить и полученные фрагменты разделить, очистить и идентифицировать. Для определения относительного содержания компонентов проводят точный количественный анализ. Далее уже нужна известная изобретательность, чтобы собрать из всех фрагментов единое целое и установить структуру исходных молекул.
а. Фракционирование клеток [105—110]
Исходным материалом при фракционировании может служить свежая ткань или осадок спрессованных клеток микроорганизма, обычно получаемый с помощью центрифугирования.
Ткань измельчают либо в мясорубке, либо, если нужна более мягкая обработка, в гомогенизаторе1. Клетки микробов чаще всего разрушают с помощью ультразвука или продавливанием через пресс под высоким давлением. При фракционировании очень важно подобрать нужное значение pH и состав буфера, а при выделении субклеточных органелл — осмотическое давление. Для сохранения целостности органелл часто в качестве суспендирующей среды используют 0,25 М сахарозу, к которой добавляют MgCl2, а также реагент, образующий комплекс с металлами, например этилендиаминтетраацетат (ЭДТА) (табл. 4-2). Растворимые ферменты обычно экстрагируют без добавления сахарозы, но при этом используют восстановители — глутатион (дополнение 7-Б), меркаптоэтанол или дитиотреитол (разд. 3.3.а).
Полученный сырой гомогенат процеживают и обычно центрифугируют непродолжительное время, чтобы удалить фрагменты клеток и прочие «осколки». Клеточные органеллы, как правило, отделяют центрифугированием [106, 107]. Одна из методик подобного рода состоит в следующем. Гомогенат в 0,25 М сахарозе (изотоничной по отношению к большей части клеток) центрифугируют в течение 10 мин при 600—1000 g, что позволяет осадить ядра и целые клетки. Затем надосадочную жидкость центрифугируют еще 10 мин при ~ 10 000 g, в результате чего осаждаются митохондрии и лизосомы. Наконец, центрифугирование в течение часа при ~ 100 000 g дает микросомный осадок. Каждый из разделенных компонентов можно затем снова суспендировать и повторно центрифугировать, чтобы получить более чистый препарат органелл данного типа. Осажденные частицы во многих случаях растворяют с помощью химической обработки, например добавляя детергенты. Супернатант (надосадочная жидкость), остающийся после центрифугирования с максимальной скоростью, служит исходным материалом для выделения растворимых ферментов и многих низкомолекулярных соединений.
1 Наиболее широко используется гомогенизатор, представляющий собой цилиндрический патрон, внутри которого вращается довольно плотно прилегающий к стенкам стеклянный или пластиковый поршень.
б. Разделение, основанное на разной растворимости компонентов системы
Некоторые фибриллярные белки практически нерастворимы в воде, так что все остальные компоненты препарата можно удалить растворением. Растворимые белки чаще всего осаждают из водных растворов путем высаливания, которое сводится к добавлению большого количества соли, например сульфата аммония. Разные белки осаждаются при разных концентрациях соли, поэтому можно отделить фракцию белков, осаждающихся в заданном интервале концентраций, а затем очищать эту фракцию дальше. Методы, основанные на таком поэтапном осаждении, широко используются как первый шаг очистки, поскольку они позволяют получать сразу большие количества материала.
РНК чаще всего получают обработкой препарата фенолом, насыщенным водой, в котором осаждаются все белки. При определенных условиях иногда удается удалить и ДНК. Последовательные стадии осаждения и экстрагирования позволяют отделить РНК от полисахаридов (в водном слое), ДНК (если она имеется) и других компонентов [111 —113]. Нуклеиновые кислоты можно отделить от белков, удаляя последние с помощью протеолитических ферментов.
Некоторые углеводы, например целлюлоза и гликоген, устойчивы к кипячению в щелочи, и это позволяет удалить все другие компоненты смеси. Липиды экстрагируют из тканей неполярными растворителями [114], например смесью СНСl3 и СН3ОН в соотношении 2:1.
в. Разделение, основанное на распределении соединений между разными фазами
Многие наиболее важные методы разделения основаны на многократном распределении соединений между двумя разными фазами, из которых хотя бы одна обычно является жидкой. Малые молекулы можно разделить с помощью противоточного распределения, когда препарат многократно уравновешивается между двумя жидкими фазами, причем одна из фаз более полярна, чем другая. После каждого уравновешивания с помощью специального устройства «противоточным» образом вводятся новые порции обеих жидкостей [115].
В качестве одной из фаз можно использовать порошок или мелкие частицы; ими заполняют вертикальную колонку или наносят их тонким слоем на стеклянную пластинку. Все эти методы называются хроматографическими — термин, предложенный М. Цветом, который впервые в 1903 г. описал разделение пигментов, присутствующих в листьях растений (хлорофилла и каротинов). Пропуская пигменты, растворенные в неполярных растворителях (например, гексане), через колонку с окисью алюминия или другими адсорбентами, М. Цвет обнаружил, что смесь разделяется на окрашенные полосы, которые перемещаются вдоль колонки по мере прохождения растворителя. Непрерывно пропуская растворитель через колонку, можно элюировать отдельные пигменты в чистом виде. Описанный метод широко применяется и сейчас и носит название адсорбционной хроматографии. При этом пигменты не растворяются в твердом материале, а адсорбируются на его поверхности.
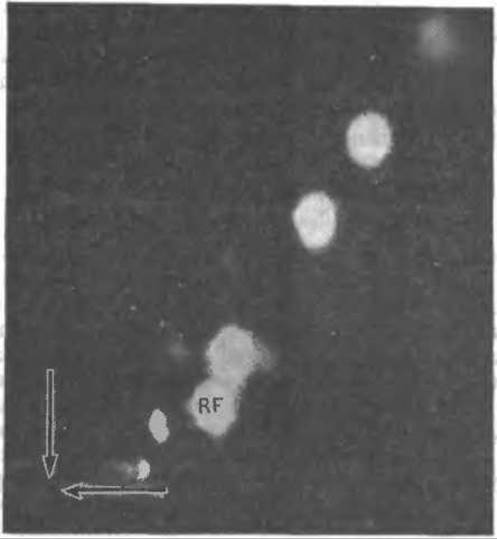
РИС. 2-34. Двумерная тонкослойная хроматография (в силикагеле) смеси флавинов, образовавшихся при облучении витамина рибофлавина. Появление светлых пятен на снимке обусловлено флуоресценцией соединений в ультрафиолетовом свете. Какое-то количество рибофлавина сохраняется в первоначальном виде (пятно, обозначенное RF). Стрелки указывают место нанесения образца. Для разделения в первом направлении использована смесь уксусная кислота/2-бутанон/метанол/бензол, во втором — смесь н-бутанол/уксусная кислота/вода [147].
Часто в качестве материала, которым наполняют колонки, используют силикагель, содержащий большое количество воды; разделение компонентов в этом случае происходит за счет их распределения между водной фазой, иммобилизованной силикагелем, и водой, протекающей через колонку. Для разделения липидов применяют колонки с обращенной фазой; их наполняют силикагелем, окисью алюминия или другим инертным материалом, пропитанным неполярной жидкостью. В роли подвижной фазы в этом случае выступает более полярный растворитель.
Хроматография на бумаге основана на использовании в качестве иммобилизованной фазы высококачественной фильтровальной бумаги, адсорбирующей воду. В последнее время широкое распространение получила тонкослойная хроматография; вместо бумаги здесь используется тонкий слой силикагеля, нанесенный на стеклянную пластинку. Этот метод гораздо удобнее хроматографии на бумаге, поскольку дает более быстрое и качественное разделение (рис. 2-34). Для разделения летучих веществ применяется газовая хроматография, основанная на установлении динамического равновесия между газовой и неподвижной фазами при сравнительно высоких температурах; область применения этого метода расширяется в связи с возможностью образования летучих производных разделяемых соединений (например, метиловых эфиров или триметилсилильных производных сахаров).
Весьма важное место принадлежит аффинной хроматографии [116, 117]. Она основана на использовании особых адсорбентов, специфически взаимодействующих с макромолекулами и избирательно удерживающих данный вид макромолекул (отсюда и название метода). Примером, о котором уже говорилось в разд. Г.10, служит адсорбция комплементарных фрагментов молекулы нуклеиновой кислоты на иммобилизованной ДНК. Аффинная хроматография используется также для очистки ферментов, антител и других белков, способных прочно связываться со специфическими малыми молекулами.
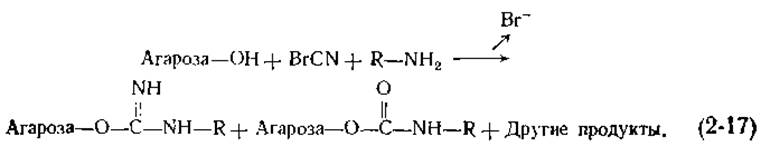
Открытый характер структуры в гелях (рис. 2-18), образуемых производными агарозы, делает крупнозернистые порошки этих гелей удобной твердой основой для приготовления адсорбентов. Гидроксильные группы агарозы часто модифицируют присоединением к ним аминов. Вначале агарозу обрабатывают бромцианом (Вr—C ≡ N) в щелочной среде, «активируя» тем самым углеводную цепь (химическая сторона этого вопроса обсуждается в работе [118]), а затем добавляют амины. Полная реакция описывается следующим уравнением:
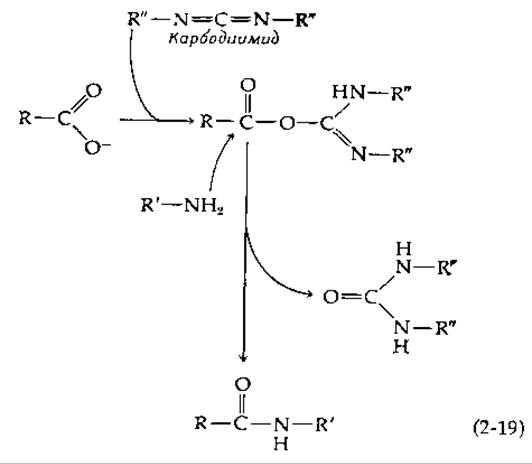
Таким образом можно приготовить адсорбенты, содержащие множество разных R-групп. Кроме того, присоединив к активированной агарозе диамин [R = (CH2)n—NH2], с полученной в результате ω-аминоалкилагарозой можно далее связать другие соединения. Для этого используется реакция с растворимым в воде карбодиимидом;
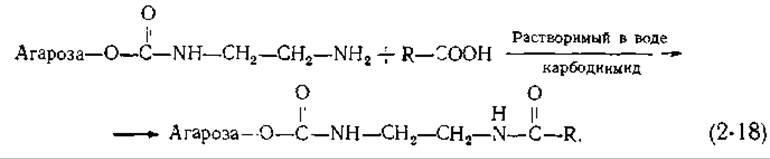
Карбодиимиды широко применяются в лабораторной практике для образования амидных и фосфодиэфирных связей. Процесс образования амида, фигурирующего в уравнении (2-18), можно изобразить в виде схемы (2-19). Для реакций, протекающих в неводной среде, часто используют дициклогексилкарбодиимид (R"= циклогексил; см схему), однако для последующего присоединения каких-либо групп к сахарозе лучше брать растворимый в воде реагент, например 1-этил-3-(диметиламинопропил)-карбодиимид [119].
Помимо описанных выше, известно также много других способов приготовления адсорбентов для аффинной хроматографии.
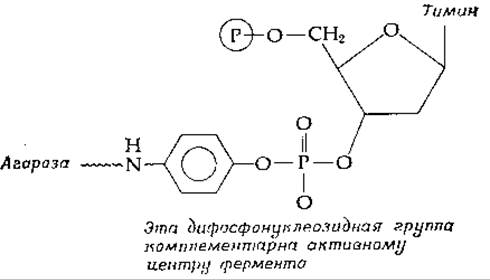
Примером применения аффинной хроматографии для очистки ферментов может служить выделение стафилококковой нуклеазы (фермента, гидролизующего ДНК) с помощью хроматографии на колонке с агарозой, содержащей следующую труппу [120]:
г. Разделение, основанное на размерах молекул
Диализ [121] и ультрафильтрация [122] основаны на применении в качестве полупроницаемого барьера тонкой мембраны (например, из ацетата целлюлозы — целлофана), имеющей поры диаметром 1—10 нм (чаще всего 5 нм). Малые молекулы такая мембрана пропускает, а крупные задерживает. Диализ определяется диффузией, и его можно ускорить перемешиванием раствора, а скорость фильтрования зависит от разности давлений по разные стороны мембраны. Более сложный характер имеет гель-фильтрация [123]. Колонку наполняют материалом типа декстрана с большим числом поперечных сшивок (сефадекс1) обычно он имеет вид спрессованных мягких шариков. Шарик представляет собой трехмерную сеть из углеводных цепей (рис. 2-18). Пространство между цепями (оно зависит от числа сшивок, образующихся в геле при его химической обработке) недостаточно для проникновения туда крупных молекул, но в нем вполне могут задерживаться малые молекулы. При пропускании через такую колонку смеси разных молекул малые молекулы диффундируют внутрь частиц геля и задерживаются там, а крупные проходят беспрепятственно (рис. 2-35). Сефадекс G-25 не задерживает ничего, кроме низкомолекулярных солей и соединений типа простых циклических сахаров. Сефадекс G-200 с гораздо меньшим числом поперечных сшивок применяется для разделения макромолекул, мол. вес которых лежит в интервале 5000—200 000.
1 Коммерческое название продукта, производимого фирмой Pharmacia Fine Chemicals, Uppsala, Sweden.
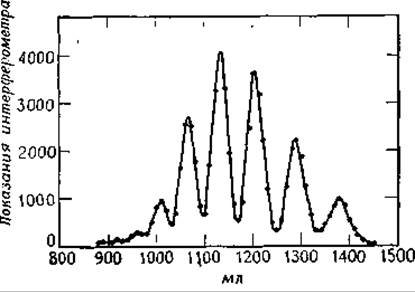
РИС. 2-35. Разделение олигосахаридов с помощью гель-фильтрации. Сахара, растворенные в дистиллированной воде, пропускали через колонку, заполненную сефадексом G-25. Пики (справа налево) соответствуют глюкозе, целлобиозе, целлотрнозе и т. д. (Flodin Р., Aspberg К., Biol. Struct. Funct. 1st., Proc. IUB/IUBS Int. Symp., 1960, 1, 345, 1961.)
д. Центрифугирование
Самые разнообразные методы разделения связаны с использованием препаративных центрифуг [124, 125]. Скорость седиментации молекул в центробежном поле зависит от формы и размеров молекул. Каждая молекула характеризуется своей константой седиментации s, выражаемой в единицах Сведберга, S. Когда же в центрифуге устанавливается седиментационное равновесие, то главным фактором, определяющим, где в итоге окажутся частицы, становится их плотность.
Для грубого предварительного разделения клеточных фрагментов достаточно кратковременного последовательного центрифугирования при нескольких разных скоростях (с разным центробежным ускорением) (разд. 3.1. а). Однако, чтобы получить максимально чистые препараты органелл или молекул, используют центрифугирование в градиенте плотности. Например, для разделения РНК на несколько фракций, различающихся по константам седиментации, сначала в пластмассовой центрифужной пробирке создают градиент концентраций раствора сахарозы (от 25% на дне до 5% на поверхности). Затем сверху аккуратно наслаивают препарат РНК и проводят центрифугирование с очень высокой скоростью в течение нескольких часов. Препарат РНК разделяется на ряд медленно седиментирующих резких полос, стабилизируемых градиентом сахарозы. Затем пробирку прокалывают снизу и собирают фракции по каплям в пробирки с помощью коллектора фракций. Далее определяют положение каждой фракции и содержание в ней РНК.
При достаточно большом времени центрифугирования частицы достигают в градиенте плотности равновесных положений. Именно тащим образом в соответствии с их плотностью в градиенте сахарозы разделяют клеточные органеллы (гл. 1, разд. Б.6). Макромолекулы (например, ДНК) чаще всего разделяют в градиенте CsCl [126]. В плотных солевых растворах градиенты достаточно устойчивы, и для лучшего разделения применяются очень сильные центробежные поля. В результате можно отделить одноцепочечную ДНК от двухцепочечной или разделить препараты ДНК с разным GC-содержанием. Последнее основано на различиях в плавучей плотности р таких ДНК в растворе CsCl, которая меняется с GC-содержанием в соответствии со следующим приближенным уравнением:
р= 1,660 + 0,098∙(мольная доля GC-пap).
е. Электрический заряд
В основе ряда методов разделения молекул лежит различие в суммарном заряде, который они несут при данном pH среды. Этот суммарный заряд для многих соединений легко оценить по числу содержащихся в них кислых и основных групп. Предположим, что нам известны значения рКа каждой группы и степень ее диссоциации при данном pH. При некотором pH, называемом изоэлектрической точкой, суммарный заряд молекулы становится равным нулю — в электрическом поле она не перемещается. При любом другом значении pH молекула будет двигаться к аноду (+) или катоду (—).
Электрофорез, процесс разделения молекул, основанный на разной скорости перемещения их в электрическом поле, проводят самыми разными способами. Очень небольшое количество раствора, содержащего смесь белков (например, белков сыворотки крови), наносят в виде тонкой полоски на лист фильтровальной бумаги или ацетата целлюлозы. Лист насыщают буфером и пропускают через него электрический ток. Напряжения в несколько сот вольт достаточно для разделения белков сыворотки в течение 1 ч. Для ускорения процесса и снижения диффузии низкомолекулярных веществ широко используют высоковольтный электрофорез. Прикладываемое напряжение составляет в этом случае 2—3 тыс. вольт. Образец постоянно охлаждают с помощью термостатируемых пластин; иногда для той же цели всю систему погружают в сосуд с керосином. Электрофоретическое разделение больших количеств материала проводится в плоских лотках, заполненных крахмальным или каким-либо другим гелем. Одним из наиболее распространенных и чувствительных методов разделения белков является электрофорез в колонке, заполненной полиакриламидным гелем. Этот метод, в настоящее время сильно усовершенствованный, позволяет проводить разделение молекул одновременно и по размеру, и по электрическому заряду; его называют методом электрофоретического молекулярного сита [127, 128].
При изоэлектрическом фокусировании в вертикальной колонке между анодом и катодом электрохимически создается градиент pH. Градиент pH стабилизируется градиентом плотности, чаще всего сахарозным; вся система тщательно термостатируется. Белки в колонке движутся в направлении, соответствующем их изоэлектрической точке, где «фокусируются» в виде очень узкой полосы. Две соседние полосы могут быть разделены интервалом всего лишь в 0,01 единицы pH. Считается, что изоэлектрические точки белков в обычных экспериментальных условиях близки к их изоионным точкам, т. е. к тем значениям pH, при которых белки остаются изоэлектрическими в условиях полного отсутствия добавленного электролита [129].
Чрезвычайно важным методом разделения, связанным с наличием электрических заряженных групп, является ионообменная хроматография. Обычно применяются водные растворы смесей и колонки, наполненные ионообменной смолой — пористым веществом, содержащим связанные ионные группы, например —SO3, —СОО-, —NH+3, или четвертичные атомы азота. Для разделения малых молекул, как правило, берут синтетические смолы, основу которых составляет полистирол с поперечными сшивками. Для крупных молекул больше подходят производные целлюлозы или поперечносшитых декстранов (сефадекс). Соединения с положительно заряженными группами, например аминокислоты в кислом растворе, наносят на колонку с катионообменной смолой, скажем дауэкс 50, которая содержит диссоциированные группы сульфоновой кислоты. Адсорбированные аминокислоты элюируют раствором НСl или буфером с возрастающим значением pH. Процесс такого типа лежит в основе автоматического количественного анализа смесей аминокислот, образующихся в результате гидролиза белков или пептидов (рис. 2-36). На сульфонат-замещенном полистироле можно разделять также пуриновые и пиримидиновые основания. Отрицательно заряженные нуклеотиды обычно разделяют на смолах, содержащих четвертичные основания [130]. Разработан вариант этого метода для разделения очень малого количества материала; например, можно количественно определять содержание каждого из нуклеотидов в препарате митохондрий из печени крысы, содержащем 5—8 мг белка [131].
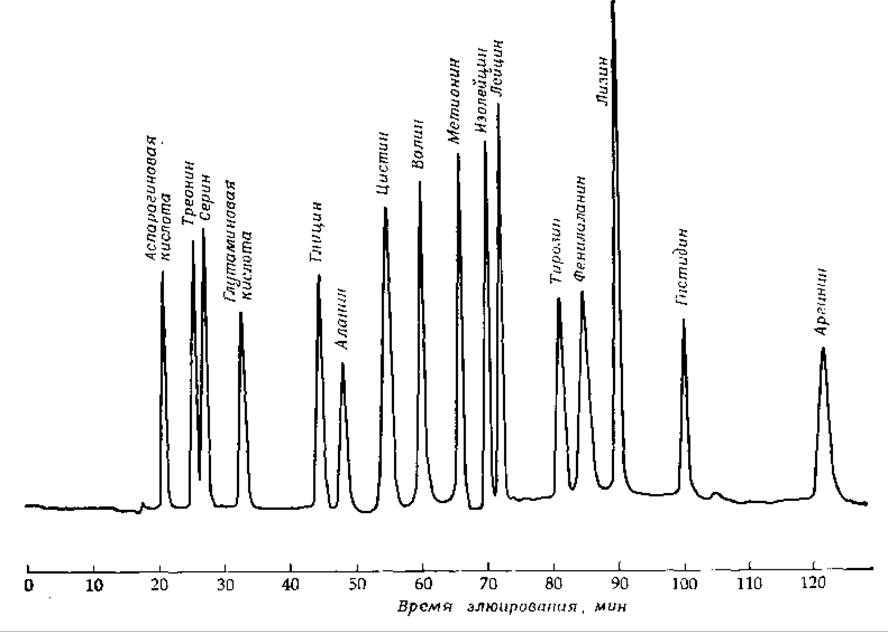
РИС. 2-36. Результат ионообменной хроматографии смеси аминокислот в аминокислотном анализаторе. Смесь аминокислот (по 0,5 нмоль каждой) нанесли на колонку диаметром 0,9 см и длиной 58 см, заполненную мелкими частицами полистирол-сульфоната (Durrum тип DC-4A), и элюировали ее цитратным буфером при трех значениях pH. последовательно возраставших от 3,5 до 6,4. Проходящий через колонку раствор обрабатывали флуорескамином (стр. 180) и затем непрерывно регистрировали флуоресценцию образовавшегося продукта. (С любезного разрешения Durrum Chemical Corp.)