ПІДРУЧНИК ДИТЯЧА ГІНЕКОЛОГІЯ - 2013
Розділ 7. АНОМАЛІЇ РОЗВИТКУ СТАТЕВИХ ОРГАНІВ У ДІВЧАТ. КЛІНІКА, ДІАГНОСТИКА ТА ЛІКУВАННЯ
ЕМБРІОГЕНЕЗ СТАТЕВОЇ СИСТЕМИ
Розвиток статевих залоз в ембріогенезі зумовлений набором статевих хромосом, які утворюються після запліднення яйцеклітини. Каріотип 46ХХ визначає розвиток яєчника, а каріотип 46ХY — яєчок. Первинне статеве диференціювання — це процес розвитку статевих гонад, який починається на 6-7-му тижні ембріонального періоду. Під впливом транскрипційних факторів WТ1 (туморосупресор пухлини Вільмса), стероїдного фактора 1 (SF1) і DAX1 формується первинна гонада. Вона складається з кортикальної та медулярної частин. При двох Х-хромосомах її кортикальна частина трансформується у яєчник. Y-хромосома сприяє розвитку медулярної частини первинної гонади в яєчко. Адже процес статевого диференціювання більш специфічний і знаходиться під контролем генів: SRY, SOX9, DSS. Ген SRY виконує основну роль у статевій диференціації та може викликати розвиток протилежної статі. До 8-го тижня гестації яєчки здатні секретувати статеві гормони, які і визначають розвиток зовнішніх статевих органів. Диференціація зовнішніх статевих органів за чоловічим типом знаходиться під впливом тестостерону, який секретується клітинами Лейдіга ембріона. Поряд з тестостероном важливе значення у диференціації статевих ознак має гормон, який пригнічує розвиток мюллерових проток і секретується клітинами Сертолі, — антимюллерів гормон (АМГ). Якщо є нормальна взаємодія АМГ і рецепторів до нього в ембріона чоловічої статі, мюллерові протоки інгібуються. Таким чином, для нормального чоловічого фенотипу потрібна Y-хромосома з геном SRY, клітини Лейдіга та Сертолі, секреція ними тестостерону й АМГ, наявність у клітинах-мішенях рецепторів до цих гормонів. Дефект у кожній ланці цього процесу призводить до формування організму за жіночим типом. За наявності каріотипу 46ХХ примордіальні клітини розвиваються в яєчник. Генами, що інгібують розвиток первинної гонади в яєчко, є DAX1, SOX3, Wnt4. Трансформація первинної гонади в яєчник відбувається на 17-20-му тижні ембріогенезу, коли примордіальні клітини перетворюються в овоцити (аналог клітин Сертолі). Їх кількість досягає максимуму (6,7 млн) на 5-му тижні гестації. До 7 років життя дитини їх кількість зменшується до 300 тис. За наявності лише однієї Х-хромосоми (45X0) овоцити піддаються дегенерації ще до народження дитини, друга Х-хромосома необхідна для підтримки маси яєчника та його подальшого розвитку. Мюллерові протоки утворюють репродуктивні органи жінки: матку, маткові труби та верхню третину піхви. Дві третини піхви утворюються з урогенітального синуса. Таким чином, формування жіночих статевих органів перебігає без активної участі функціонування яєчників.
Диференціювання первинної гонади в яєчник — це пасивний процес, який індукується специфічними молекулами, що відповідають H-Y-антигену в особи чоловічої статі. У диференціюванні яєчників провідну роль відіграють Х-хромосоми.
Аномалія одного або кількох детермінант статі, поломка комплексного механізму, який «запускає» статевий розвиток, можуть призвести до анатомічних і функціональних відхилень від норми, різних клінічних форм порушення статевого диференціювання. Різноманітні хромосомні аберації, генні мутації, які сприяють порушенням гормонального балансу або зміні рецепції гормонів у ембріональному періоді, можуть бути причинами вроджених аномалій статевого розвитку. Поряд із хромосомними абераціями (структурними та кількісними), генними мутаціями вроджені порушення статевого диференціювання можуть бути зумовлені також різноманітними ембріональними чинниками (інтоксикація, інфекція, травма, лікарські засоби), порушенням гормонального балансу у вагітної в критичні періоди для формування статевого тракту.
Тип порушення статевого диференціювання залежить від причини і часу його виникнення в онтогенезі. На ранніх етапах ембріогенезу (6-10 тиж.) виникає агенезія гонад. Якщо з будь-яких причин порушується процес диференціювання (формування гонад), то може розвинутися організм без статевих залоз і функціональних елементів (агенезія гонад). Найчастіше агенезія гонад є наслідком патології статевих хромосом. Інколи причиною агенезії гонад бувають інші ушкоджувальні чинники (інтоксикація, інфекція, радіація), які перешкоджають формуванню гонад. У таких випадках виявляють нормальний для жінок і чоловіків набір статевих хромосом. Незалежно від причини, що викликала агенезію гонад, клінічна картина багато в чому подібна. Замість гонад у дівчат знаходять смужки сполучної тканини, які не містять функціональних елементів. У подальшому розвивається жіночий фенотип з вираженим гіпогонадизмом, і як наслідок — відсутні фетальні гонади й автономна тенденція будь-якого плода до розвитку за жіночим, «нейтральним» типом. Отже, агенезія гонад є найбільш ранньою патологією статевого формування, до якої належать синдром Шерешевського—Тернера та «чиста» агенезія гонад.
У пізні терміни ембріогенезу виникає патологія статевого формування, яка називається дисгенезією гонад. Дисгенезія гонад — узагальнене поняття, що включає низку синдромів, зумовлених порушеннями ембріонального розвитку гонад у результаті хромосомних аберацій, генних мутацій або ембріотоксичних чинників. До них належать синдром дисгенезії яєчників і синдром дисгенезії яєчок. У багатьох випадках синдром дисгенезії яєчників визначає мозаїчний каріотип 45 Х0/46 XX, що перешкоджає нормальному диференціюванню яєчника. При нормальному жіночому каріотипі не є виключенням генна мутація, можлива дія інших ушкоджувальних чинників, які сприяють формуванню неповноцінного, дисгенетичного яєчника. Фенотип у дівчат із синдромом дисгенезії яєчників завжди жіночий, а неповноцінність яєчника проявляється лише в пубертатному періоді більш або менш вираженим гіпогонадизмом.
Порушення формування статевого тракту може бути пов’язане з недорозвиненням і дисгенезією яєчок, причини їх аналогічні. Дисгенетичні яєчка не забезпечують регрес парамезонефральних проток і нормальну маскулінізацію зовнішніх геніталій, що спричинює розвиток дериватів парамезонефральних проток.
Генетично зумовлені порушення статевого диференціювання при синдромі тестикулярної фемінізації відбуваються в період ембріогенезу. Внаслідок порушення специфічної реакції органів-мішеней на андрогени розвивається жіночий фенотип у плода генетично і гонадної чоловічої статті. Внутрішні статеві органи у пацієнток чоловічі, а зовнішні — жіночі. При неповній формі синдрому спостерігається незначна маскулінізація зовнішніх геніталій. Одним із хромосомних синдромів, що перебігає з порушеннями статевого розвитку, є синдром Шерешевського—Тернера, типова клінічна картина якого проявляється при каріотипі 45Х0. Існують такі варіанти мозаїцизму: 45Х0/46ХХ; 45X0/46XY; 45Х0/47ХХХ, значно рідше — 45X0/47XXY. Маса тіла у дітей із синдромом Шерешевського—Тернера при народженні є меншою за норму. У новонароджених виявляють лімфатичний набряк стоп, гомілок, кистей рук, складки шкіри на шиї. Затримка росту проявляється в будь-якому віці, в окремих випадках — від народження. Відставання в рості особливо помітне в пубертатному періоді. Зріст рідко досягає 150 см.
При огляді привертає увагу загальна диспластичність: діти коренасті (низько посаджена голова, коротка шия, діжкоподібна грудна клітка з широко розставленими сосками і втисненням у ділянці груднини — «щитоподібна грудна клітка»). Для пацієнток із синдромом Шерешевського—Тернера є характерними: коротка шия з широкою основою, низька межа росту волосся на шиї ззаду, крилоподібні шкірні складки від потилиці до надпліччя, так звана «шия сфінкса». Виділяють такі особливості черепа обличчя: мікрогнатія, ретрогнатія, птоз, епікант, деформовані та низько розташовані вуха. Мозковий череп є відносно більшим, ніж череп обличчя. У деяких випадках спостерігаються вальгусна девіація ліктьових і колінних суглобів, укорочення метакарпальних і метатарзальних кісток, деформація нігтів, що глибоко сидять у нігтьовому ложі. На шкірі — численні пігментні плями, вітиліго. Спушені кінчики пальців (подушечки). Уроджені вади розвитку внутрішніх органів, такі як коарктація аорти, незарощення міжшлуночкової перегородки й артеріальної протоки, стеноз аорти, легеневої артерії, підковоподібна нирка, подвійні миски і сечоводи, ротація, гіпоплазія нирки. Інтелект знижений.
Статевий інфантилізм проявляється у пубертатному періоді: відсутні вторинні статеві ознаки, молочні залози не розвиваються, вторинне оволосіння відсутнє або незначне, недорозвинуті соромітні губи, піхва та матка. Слизова оболонка вульви і піхви суха, епітеліальний покрив стоншений. Пацієнтки із синдромом Шерешевського—Тернера звертаються по медичну допомогу переважно у зв’язку з низькорослістю. При УЗД матка інфантильна, гіпоплазована, придатки у вигляді тяжів. Рентгенологічне дослідження кистей (кістковий вік відстає від норми на 2-4 роки) — гіпертрофічний остеопороз. На краніограмі — гіперпневматизація пазух основної кістки. Пневмопельвіограма показує різко гіперплазовану матку і тяжі в місцях розташування яєчників. Підвищення гонадотропної активності, особливо ФСГ, у пубертатному періоді (9-13 років), максимальна функціональна активність — 16-17 років.
Секреція ЛГ має такий же характер, але його рівень зазвичай становить 1/2-1/6 рівня ФСГ. Різке збільшення ЛГ і ФСГ із частими хаотичними десинхронізованими коливаннями, зберігається зворотний зв’язок у системі гіпофіз — гонади. Екскреція естрогенів низька, вагінальні мазки атрофічного типу. Незначне підвищення рівня СТГ, можлива резистентність тканин до тиреоїдних гормонів. Знижена толерантність до глюкози, глікемічна крива сумнівна або діабетоїдна. За допомогою цитогенетичного дослідження виявляють каріотип 45X0 або 45Х0/46ХХ. Статевий хроматин відсутній або різко знижений. Хвора потребує гормонального лікування.
Класичний варіант синдрому Клайнфельтера є проявом тріади симптомів: гіпогонадизму, гінекомастії та дисгенезії сім’яних канальців яєчок. Причиною даного стану є одна або більше зайвих Х-хромосом у каріотипі, тобто хромосомний набір 47XXY або 48XXXY. Інколи виявляють хромосомний мозаїцизм типу 47XXY/46XY або 47XXY/46XX; 48XXXY/46XY або 47XXX/47XXY. Кожна зайва Х-хромосома посилює інтелектуальну недостатність, а за умов мозаїцизму зовнішні прояви синдрому можуть бути невираженими або стертими. Основним проявом аномального набору статевих хромосом є дефект функції інтерстиціальних клітин і, як наслідок, асперматогенез. Порушення функції інтерстиціальних клітин проявляється ознаками загальної андрогенної недостатності, з одного боку, і місцевої гіалінізації сім’явивідних канальців — з другого. Клінічна діагностика синдрому Клайнфельтера в період новонародженості неможлива, тому що жодних характерних особливостей при обстеженні новонароджених хлопчиків з аномальним набором статевих хромосом 47XXY не виявляють. При дослідженні мазків слизової оболонки щоки визначають хроматинпозитивні інтерфазні ядра. Обов’язкова ознака захворювання — гіпоплазія гонад — у допубертатному періоді визначається зрідка. У деяких випадках у цьому періоді проявляються ознаки затримки розумового розвитку, що може бути підставою для дослідження статевого хроматину. Попередній діагноз захворювання на основі клінічних даних можливий лише у препубертатному або пубертатному періоді. Незважаючи на нечисленність ознак, клініка синдрому відрізняється достатньо вираженим поліморфізмом. Залежно від соматичного розвитку, виділяють такі типи синдрому Клайнфельтера: диспластичний — худорлявий тип, псевдом’язовий, з так званим євнухоподібним відкладанням жирової тканини на стегнах, грудях і нижній частині живота. При євнухоподібному складі хроматинпозитивні пацієнти зазвичай є високими на зріст, що зумовлено довгими ногами. Зафіксована схильність до вузьких плечей при відносно широкому тазі, плоска і вузька грудна клітка, слабкий розвиток мускулатури, сутуловатість. У підлітків спостерігається синюшність кистей і стоп, їх підвищена пітливість.
Гінекомастія розвивається у препубертатному і пубертатному віці (з 12 до 16 років), як правило, з обох боків. Тканина молочної залози складається з проток, навколо яких переважає розвиток щільної фіброзної тканини, зазвичай залози не секретують. Розвиток гінекомастії зумовлений підвищеним рівнем естрогенів інтерстиціальними клітинами тестикул. Зовнішні статеві органи — чоловічого типу. Яєчка опущені в мошонку, щільні, дуже маленькі — 1,5 см (при нормі 5 см), внутрішні статеві органи значно менших розмірів порівняно з нормою. Оволосіння на обличчі незначне, водночас є характерними низька межа росту волосся на лобі, рідке оволосіння під пахвами, на лобку — за жіночим типом (вочевидь, зумовлено це загальною кількісною та якісною недостатністю андрогенів, а також зниженою чутливістю тканин до андрогенів). Характерним є також високий рівень гонадотропінів.
Синдром тестикулярної фемінізації передається через патогенні гени жінок-носіїв, унаслідок чого відмічається слабке статеве оволосіння, пізній початок менструацій. Його розвиток пояснюється недостатньою активністю або нестабільністю ферменту 5-α-редуктази, що відповідає за утворення важливого метаболіту тестостерону 5-α-дигідротестостерону, який забезпечує розвиток зовнішніх геніталій за чоловічим типом. При даному синдромі знижені або повністю відсутні рецептори до андрогенів у клітинах деяких тканин. За відсутності реакції тканин на статевий гормон тонічна секреція гонадотропінів підвищується.
При тестикулярній фемінізації різко підвищений вміст ЛГ в крові. Гонадотропіни, що взаємодіють зі специфічними рецепторами, розташованими всередині клітинних мембран, не тільки реагують із рецепторами білка, але й впливають на їх утворення. Значна кількість ЛГ гальмує власні рецептори. Незважаючи на ареактивність тканин до андрогенів, відбувається нормальна маскулінізація центрів, що регулюють секрецію гонадотропінів. Доведено, що в основі патогенезу лежить генетично зумовлена ареактивність тканин до андрогенів при збереженій чутливості до естрогенів. Фетальні яєчка мають антимюллерові властивості, що призводить до атрофії парамезонефральних проток, унаслідок чого у дівчат відсутні матка, маткові труби і верхня третина піхви.
Внутрішні геніталії чоловічі, передміхурова залоза відсутня, наявний сліпий піхвовий відросток урогенітального синуса. При повній ареактивності тканин до андрогенів або порушенні біосинтезу тестостерону маскулінізації зовнішніх геніталій не відбувається і вони зберігають жіночу, нейтральну будову. Явища фемінізації у пубертатному періоді пояснюються підвищеною продукцією естрогенів яєчками внаслідок посиленої стимуляції гонадотропінами.
Залежно від вираженості естрогенної та андрогенної дії виділяють дві форми тестикулярної фемінізації. Для повної форми характерна абсолютна відсутність чутливості до андрогенів, у результаті чого дівчата мають жіночий фенотип і правильну будову тіла, розвиваються молочні залози. Статеве оволосіння відсутнє або незначне, тимчасом як волосся на голові густе, зовнішність приваблива, жіноча. Неповна форма характеризується частково збереженою чутливістю органів-мішеней до андрогенів, що виражається в незавершеній маскулінізації зовнішніх геніталій, яка проявляється при народженні. У пубертатному періоді проявляються чоловічі риси, інтерсексуальна будова тіла. Основними ознаками синдрому тестикулярної фемінізації є генетична і гонадна чоловіча стать, відсутність матки, маткових труб, верхньої третини піхви, незавершена маскулінізація зовнішніх геніталій, розвиток фемінізації у пубертатному періоді. Відсутні статеве оволосіння, менструації. Цитогенетично: статевий хроматин негативний, каріотип чоловічий — 46XY. Рівень базального тестостерону — в межах вікової норми для хлопчиків. Рівень базального естрадіолу перевищує вікову норму для хлопчиків, а в деяких випадках наближається до вікової норми дівчаток. Функціональна проба з хоріонічним гонадотропіном неоднозначна. Підвищений рівень ЛГ. Зростання гонадотропної активності спостерігається в 12-13 років. У 13-14 років збільшується рівень тестостерону й естрадіолу. До 16-17 років рівень тестостерону прирівнюється до вікової норми хлопчиків, а рівень естрадіолу підвищується.
Синдром гермафродитизму
Це група захворювань, проявом яких є двостатева структура зовнішніх геніталій.
При справжньому гермафродитизмі, або синдромі справжньої двостатевості, каріотип 46ХХ, рідше 46XY, або його поєднання. Фенотип може бути чоловічим або суто жіночим. Зовнішні геніталії двостатеві. Латерально складка з одного боку утворює половину мошонки і в ній яєчко, з протилежного — велику соромітну губу. В усіх хворих виявляють матку, інколи маткові труби. Визначають змішану гонаду. Діагноз установлюють на підставі двостатевих гонад, наявних внутрішніх жіночих органів на фоні вторинних чоловічих ознак.
Лікування полягає у формуванні особи як жінки за допомогою реконструктивної операції гонад із призначенням естрогенів, що сприяє набуттю жіночих рис.
Порушення статевого диференціювання можуть бути зумовлені не тільки хромосомними абераціями, але й генними мутаціями і різноманітними ембріотоксичними чинниками. У практичній діяльності трапляється значна кількість синдромів, що перебігають з тими або іншими ознаками порушень статевого розвитку. Відомо чимало синдромів, у основі яких лежать деякі прояви порушень статевого розвитку.
Синдром Перрольта: глухота, туговухість будь-якого типу, аплазія, гіпоплазія, дисгенезія яєчників, гіпогонадизм; аплазія, гіпоплазія матки, однорога матка; пропорційний нанізм; атаксія; затримка інтелектуального розвитку; коротка шия; сколіоз та ін. Успадковується за аутосомно-рецесивним типом.
Синдром Вудхауза: олігофренія, глухота, гіпогонадизм, затримка інтелектуального розвитку, цукровий діабет, гіпотрихоз скальпа, аплазія та гіпоплазія брів, аплазія та гіпоплазія матки, однорога матка, аплазія, гіпоплазія та дисгенезія яєчників, гіпоплазія грудних залоз тощо. Успадковується за аутосомно-рецесивним типом.
Синдром де Груші олігофренії — нанізму — інверсії статі: затримка інтелектуального розвитку; мікроцефалія; аплазія, гіпоплазія матки, однорога матка; інверсія статі, XY дисгенез; пропорційний нанізм; гірсутизм, густі брови, великий ніс; аплазія, гіпоплазія, дисгенезія яєчників; гіпоплазія соромітних губ і т. ін. Успадковується за аутосомно-рецесивним типом.
Синдром Майнеш полідактилії — сечостатевих аномалій: аплазія, гіпоплазія нирок; аплазія, гіпоплазія матки, однорога матка; аплазія, гіпоплазія, дисгенезія яєчників; пухлини статевих залоз; пренатальна гіпоплазія тощо. Тип успадкування неуточнений.
Синдром Тібі олігофренії — ожиріння — гіпогонадизму: затримка інтелектуального розвитку; гінекомастія; гіпогонадизм; ожиріння; арахнодактилія яєчників; сутулість; Х-подібне викривлення ніг і т. ін. Успадковується за аутосомно-рецесивним типом.
Ульнарно-мамарний синдром: гіпоплазія сосків; гіпогонадизм; аплазія, гіпоплазія 4-5-го пальців кисті або стопи; мікроцефалія; аплазія, гіпоплазія матки, однорога матка; пропорційний нанізм; ожиріння; євнухоподібна статура та ін.
Синдром Хеймета глухоти — нефропатії — гіпогонадизму: глухота, туговухість; нефропатія, протеїнурія; гіпогонадизм; початок захворювання після 5-річного віку; аплазія, гіпоплазія матки, однорога матка; аплазія, гіпоплазія, дисгенезія яєчників; аменорея тощо. Успадковується за аутосомно-рецесивним типом.
Синдром Аль-Евейді гіпотрихозу — гіпогонадизму: аплазія, гіпоплазія матки, однорога матка; аплазія, гіпоплазія яєчок; гіпогонадизм; осередкова алопеція; гіпотрихоз тіла; аплазія, гіпоплазія, дисгенезія яєчників; аменорея та ін. Успадковується за аутосомно-рецесивним типом.
Синдром Бернара — Уейла неврологічних аномалій — гіпогонадизму: атаксія; дизартрія, дизлалія; гіпогонадизм; високе склепіння стопи; початок захворювання проявляється після 10-річного віку; аменорея та ін. Успадковується за аутосомно-рецесивним типом.
Синдром Фраз’є гонадного дисгенезу — нефропатії: нефропатія; аплазія, гіпоплазія, дисгенезія яєчників; інверсія статі, XY-дисгенез; аменорея тощо. Неуточнений тип успадкування.
Синдром Бенгстеда олігофренії — нанізму — гіпогонадизму: атаксія; затримка інтелектуального розвитку; мікроцефалія; гіпогонадизм; пропорційний нанізм; цукровий діабет і т. ін. Успадковується за аутосомно-рецесивним типом.
Синдром Броснека олігофренії — нанізму — інверсії статі: затримка інтелектуального розвитку: тригоненцефалія; атрезія, стеноз слухових ходів; інверсія статі, XY — дисгенезія; пропорційний нанізм; аплазія, гіпоплазія матки, однорога матка та ін. Успадковується за аутосомно-рецесивним типом.
Синдром Малоуфа кардіопатії — гіпогонадизму: птоз повік; широка, висока спинка носа та перенісся; кардіопатія; аплазія, гіпоплазія, дисгенезія яєчників; гіпогонадизм та ін. Успадковується за аутосомно-рецесивним типом.
Синдром Неумена численної гіпоплазії ендокринних залоз: аплазія, гіпоплазія гіпофіза, щитоподібної залози, надниркових залоз; аплазія, гіпоплазія, дисгенезія яєчників, яєчок. Успадковується за аутосомно-рецесивним типом.
Синдром Вейнштейна глухоти — катаракти — нанізму — гіпогонадизму: катаракта; глухота, туговухість; гіпогонадизм; нанізм; ожиріння.
Синдром Фріска аномалій обличчя — гіпогонадизму: затримка інтелектуального розвитку; енофтальм; широка спинка носа, перенісся; прогенія; гіпогонадизм; євнухоподібна статура тощо. Успадковується за аутосомно-рецесивним типом.
Синдром Фітча нанізму — аномалій кистей — гіпогонадизму: гіпогонадизм; нанізм; ізодактилія; аплазія, гіпоплазія фаланг кистей та ін. Успадковується за аутосомно-рецесивним типом.
Синдром Фамізими акромегалії — чорного акантозу: широка спинка носа, товсті губи, макрогенія; гіпогонадизм; нанізм; брахідактилія; резистентність до інсуліну. Успадковується за аутосомно-рецесивним типом.
Слід зазначити, що поширеність у популяціях вищенаведених синдромів незначна і становить 1 : 200 000 - 1 : 500 000.
Аномалії розвитку матки
Синдром Майєра — Рокитанського — Кюстнера — аплазія матки за наявності маткових труб і функціональних яєчників. Іноді на місці матки може бути невеличкий м’язовий валик. Вада поєднується з частковою або повною аплазією піхви, верхньою її частиною. Для даної патології характерна первинна аменорея. Діагноз встановлюють у період статевого розвитку, вторинні статеві ознаки виражені, фенотип жіночий. Діагностичне значення має гінекологічне обстеження, ультразвукове дослідження, діагностична лапароскопія. Лікування — хірургічне і полягає у створенні піхви.
Uterus didelphus — подвоєння матки та піхви при їх окремому розташуванні.
Uterus duplex et vagina duplex — подвоєння матки та піхви, на певній ділянці вони з’єднуються фіброзно-м’язовим прошарком.
Uterus bicornis bicollis — дворога матка з двома шийками.
Uterus bicornis unicollis — дворога матка з однією шийкою.
Сідлоподібна матка — умовна перегородка всередині матки.
Стеноз дівочої пліви
У деяких жінок отвір дівочої пліви замалий, що утруднює статеві контакти. У цих випадках дівоча пліва є товстою і фіброзною. Збільшення її отвору можна виконати за допомогою маленького розтину під місцевою анестезією. За необхідності розтини виконують на «14-, 16-, 20-ту і 22-гу годину за годинниковою стрілкою», починаючи з краю дівочої пліви та продовжуючи розріз уздовж поздовжньої осі піхви. Потім ці розрізи зашивають у поперечному напрямку швами, що абсорбуються (рис. 15).
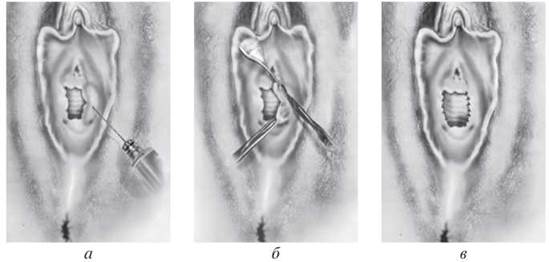
Рис. 15. Операція при стенозі дівочої пліви: а — інфільтрація hymen розчином судинозвужувального агента (0,1 % розчин адреналіну або вазопресину); б — вирізання частини hymen з вертикальним розтином; в — обшивання країв дівочої пліви окремими швами
Неперфорована дівоча пліва
Це вроджена аномалія. Пацієнтки можуть скаржитися на первинну аменорею і появу випинання у присінку піхви. При ректальному дослідженні виявляється гематокольпос. Дівочу пліву потрібно широко розкрити й ушити так, як у разі її стенозу (рис. 16). Піхву промивають антисептичними розчинами.
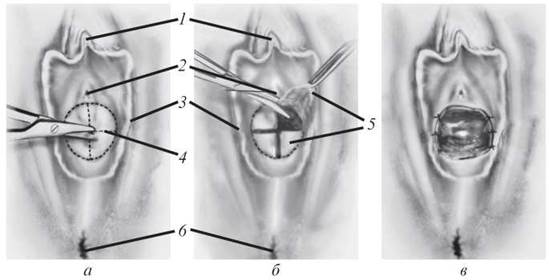
Рис. 16. Неперфорована дівоча пліва. Розтини виконують «на 3», «на 9», потім «на 12 і 6 годин»; клаптики hymen висікають, і краї рани обшивають окремими вікриловими швами № 3-0 (а-в): 1 — клітор; 2 — уретра; 3 — малі соромітні губи; 4 — лінії розтину; 5—пелюстки hymen; 6 — анус
Перегородка піхви
Перегородки бувають поперечними і поздовжніми (рис. 17).
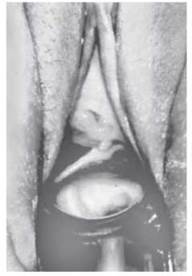
Рис. 17. Поздовжня перегородка піхви
Поздовжня перегородка піхви утворюється внаслідок неповного злиття парних мюллерових проток. Проксимальний кінець перегородки може знаходитися біля шийки матки або між подвоєними шийками. Перегородка буває повною і частковою, на короткій відстані. Інколи вона може призводити до оклюзії однієї половини піхви і створювати гематокольпос.
Більшість випадків поздовжньої перегородки піхви є безсимптомними, лише інколи вони можуть викликати біль або кровотечу під час статевого акту. При виявленні перегородки проводять хірургічне лікування. Випадки недіагностованої поздовжньої перегородки можуть призвести до тяжкої кровотечі під час пологів. Операція з її видалення є досить простою (рис. 18). Як і у разі поперечної перегородки, звертають увагу на профілактику зменшення діаметра піхви після операції.
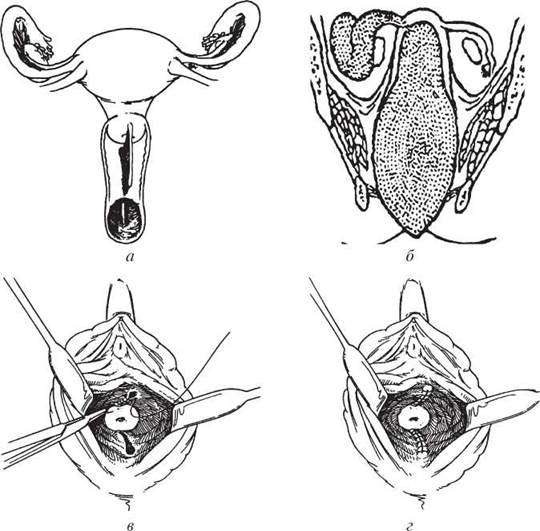
Рис. 18. Видалення поздовжньої перегородки піхви: а — локалізація перегородки; б — ексцизія перегородки за допомогою помірної тракції; в, г — відновлення дефекту слизової оболонки піхви
Поперечна перегородка піхви може локалізуватися на будь-якому рівні між дівочою плівою та шийкою матки. Перед хірургічним лікуванням слід визначити товщину перегородки. Якщо вона є неповною, її можна усунути за допомогою пальців хірурга. У деяких випадках при повній перегородці близько 1/3 піхви відсутня. Якщо ж вона є відносно тонкою, її можна відкрити і відсікти гострим шляхом (рис. 19). Під час процедури важливо зберегти діаметр піхви. У післяопераційному періоді рекомендують використовувати дилататори протягом кількох тижнів або місяців. Якщо великий сегмент піхви відсутній (понад 1 см), перегородку видаляють і використовують дилататори для профілактики стенозу піхви. Реконструкція піхви і пластика за Мак-Індо покращують результати у деяких пацієнток.
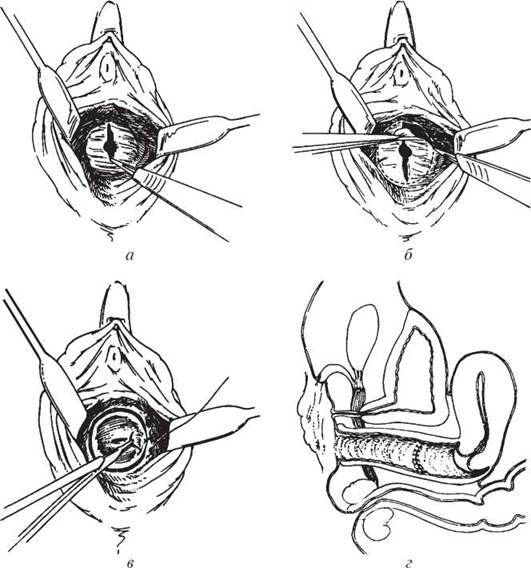
Рис. 19. Поперечна перегородка піхви: а — початковий вертикальний розтин перегородки; б — гостра дисекція перегородки від слизової оболонки піхви; в, г — відновлення дефекту слизової оболонки піхви окремими швами, що абсорбуються
Аномалії розвитку піхви
1. Агенезія піхви — первинна повна відсутність піхви.
2. Аплазія піхви — первинна відсутність частини піхви.
3. Атрезія піхви — вторинна відсутність піхви, повне або часткове її зарощення, пов’язане з внутрішньоутробним запальним процесом.
Форми атрезії:
— гіменальна, ретрогіменальна;
— вагінальна;
— цервікальна;
— маткова;
— трубна.
4. Перегородка піхви — повна, неповна.
Аномалії зовнішніх статевих органів
1. Агенезія, гіпоплазія, гіпертрофія клітора.
2. Атрезія гімена — зарощення дівочої пліви.
3. Аплазія соромітних губ, гімена.
Гінатрезії — порушення розвитку статевих шляхів: дівочої пліви, піхви або матки (рис. 20).
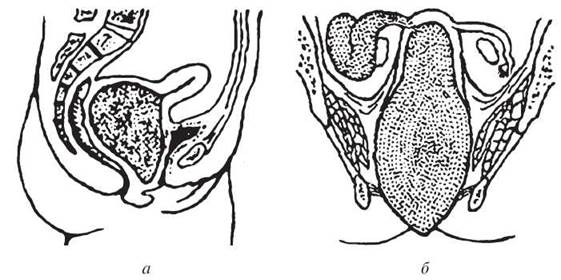
Рис. 20. Гінатрезія: а — гематокольпос при атрезії дівочої пліви; б — гематокольпос, гематометра та гематосальпінкс
Гінатрезія може бути первинною (розвивається внутрішньо- утробно внаслідок вад ембріонального розвитку) та вторинною (набутою) — внаслідок запальних процесів, перенесених у дитячому віці. У зрілому віці атрезія піхви може розвинутися внаслідок пологових травм, атрезія шийки матки — після діатермокоагуляції, атрезія порожнини матки або зрощення у ній — після надмірного вишкрібання під час аборту. При атрезії гімена з початком перших менструацій кров починає скупчуватись у піхві, розтягуючи її (haematocolpos). Дівчинка скаржиться на біль. Після закінчення такої прихованої менструації кров гемолізується, її рідка частина всмоктується, об’єм зменшується, біль припиняється до початку наступних місячних. Якщо хвора не звертається по медичну допомогу, то кров, скупчуючись усе більше, збирається у порожнині матки (haematometra), потім — у маткових трубах (haematosalpinx). У зв’язку зі здатністю піхви до розтягнення больовий синдром зазвичай з’являється при значних розмірах гематокольпоса. Гематометра та гематосальпінкс при цій патології виникають рідше.
Атрезія дівочої пліви найчастіше діагностується у період статевого дозрівання. Клінічна картина характеризується періодичним болем унизу живота у дівчаток після 12 років за відсутності менструацій (несправжня аменорея). Інтенсивність ниючого болю збільшується, при великих розмірах гематокольпоса може виникнути утруднення сечовиділення та дефекації. Вторинні статеві ознаки відповідають вікові. Під час больового нападу виникає затримка сечовипускання. При огляді присінка піхви виявляють відсутність отвору в дівочій пліві, синюшність, випинання. Ректоабдомінальним дослідженням у ділянці розміщення піхви пальпується пухлиноподібне утворення еластичної консистенції, на верхівці якого розміщується невелика щільна матка.
До періоду статевого дозрівання гінатрезії не діагностуються, після початку менструації діагностика є нескладною завдяки класичному анамнезу та клінічній картині. При атрезії гімена виконують хрестоподібний розріз дівочої пліви, краї якого ушивають окремими швами. З піхви видаляють згустки крові.
Атрезія піхви трапляється у верхньому, середньому та нижньому її відділах і має різні розміри. Кров накопичується вище місця атрезії, поступово заповнює перерозтягнутий канал шийки матки та її порожнину.
Клінічна картина: біль періодично з’являється внизу живота і в поперековій ділянці, менструація відсутня. Діагностичне значення має ультразвукове обстеження, а також зондування піхви, за допомогою якого визначається рівень атрезії. При ректоабдомінальному дослідженні пальпується тугоеластичне утворення, розміщене спереду прямої кишки. Якщо атрезія піхви ускладнюється гематометрою та гематосальпінксом, у малому тазі виявляється кілька утворень округлої форми і тугоеластичної консистенції.
Лікування оперативне. При атрезії нижньої третини піхви розшаровують тканини ділянки входу в піхву, розрізають нижній купол піхви і підшивають його слизову оболонку до входу піхви. Якщо атрезія розміщується в середній третині піхви, то розрізають тканини між нижньою та верхньою третиною піхви, а потім зшивають слизову оболонку цих ділянок. Найскладнішою є операція при атрезії верхньої третини піхви, коли відсутні склепіння, а шийка матки розміщується в клітковині малого таза. Операція є ефективною, якщо вдається знайти канал шийки матки і вшити її у верхній купол піхви.
Первинна атрезія дівочої пліви діагностується медичним персоналом або матір’ю дівчинки ще у період новонародженості. При атрезії гімена огляд зовнішніх геніталій має вирішальне діагностичне значення. На ехограмі малого таза визначається різко розширена, заповнена рідиною піхва у вигляді ехонегативного утворення, тимчасом як матка є нормальних розмірів і розташована на вершині гематокольпосу. Вважають, що спершу заповнюється розтягнута піхва, а порожнина матки не розширюється до тих пір, поки тиск нагромадженої рідини не буде перевищувати силу м’язового скорочення матки, після чого утворюються гематометра і гематосальпінкс.
Прогноз залежить від своєчасної діагностики захворювання. При тривалому перебігу хвороби і розвитку гематометри та гематосальпінксів у дівчинки в подальшому можуть бути проблеми з виношуванням вагітності, а якщо у трубах розвинулися деструктивні зміни — може виникнути їх непрохідність. Часто виникає ендометріоз внутрішніх статевих органів.
Аплазія піхви — відсутність піхви. Виникає при недостатньому розвитку нижніх (каудальних) відділів парамезонефральних (мюллерових) ходів. При цій патології матка, маткові труби і яєчники часто недорозвинені.
При аплазії піхви аменорея справжня або несправжня, статеве життя неможливе.
Діагностується на підставі даних анамнезу (аменорея) та гінекологічного обстеження хворої.
Лікування — хірургічне. Для створення піхви можна використовувати шкірний клапоть, частину сигмоподібної кишки, очеревини малого таза.
Зовнішні статеві органи утворюються із сечостатевого синуса. Якщо в процесі формування статевих органів на організм вагітної діятимуть шкідливі чинники, зокрема медикаменти, процес диференціювання статевих органів може порушитися.
Виходячи з механізму формування статевих органів, можливі такі варіанти вад розвитку матки та піхви (рис. 21, 22):
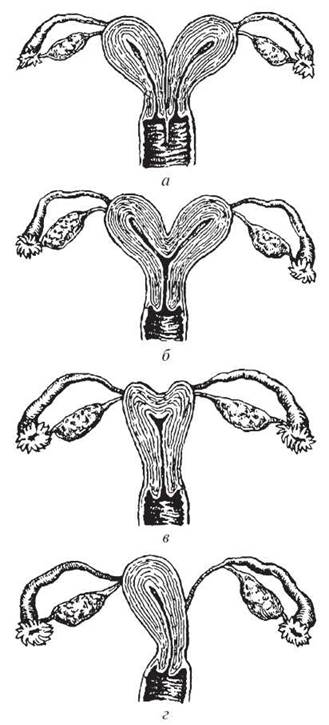
Рис. 21. Вади розвитку матки і піхви: а — дворога матка, роздвоєння шийки матки, перегородка піхви; б — дворога матка з єдиною шийкою; в — сідлоподібна матка; г — однорога матка
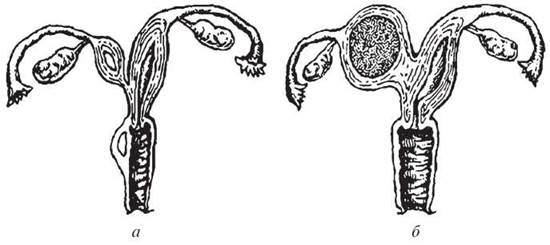
Рис. 22. Вади розвитку матки та піхви: а — рудиментарна права піхва, рудиментарний правий ріг матки; б — дворога матка, рудиментарний правий ріг матки, гематометра
1. Обидві мезодермальні протоки формуються правильно, але не зливаються між собою по всій довжині. Утворюється повне подвоєння матки та піхви: у хворої дві піхви, розділені тонкою перегородкою, в кожну піхву відкривається шийка матки, дві матки; у кожної матки — одна труба й один яєчник.
2. Обидві мезодермальні протоки формуються правильно, але їх злиття відбувається лише на певному проміжку, а інші частини матки та піхва розділені перегородкою. Варіантів такої вади може бути багато: перегородка у піхві; наявність однієї піхви, в яку відкриваються дві шийки матки; перегородка в порожнині матки; дворога матка; сідлоподібна матка.
3. Одна з мюллерових проток розвивається правильно, а друга не розвивається зовсім. Формується піхва, однорога матка з одним яєчником та однією трубою. У хворої слід обстежити сечовивідну систему, бо часто така вада поєднується з відсутністю нирки на ураженому боці.
4. Одна з мезодермальних проток розвивається правильно, друга — недостатньо, формується матка з рудиментарним рогом. Ці порожнини бувають з’єднані між собою, тому можливе настання вагітності в рудиментарному рогові. Вона розвивається як позаматкова, при перериванні відбувається значна кровотеча, що потребує хірургічного втручання.
Діагностика полягає у проведенні огляду зовнішніх статевих органів, огляді шийки матки у дзеркалах, бімануальному дослідженні. Для уточнення діагнозу інколи необхідні УЗД, зондування порожнини матки, гістеросальпінгографія або контрастна сонографія.
Лікування аномалій розвитку — хірургічне. Подвоєння матки та піхви, яке не порушує статевої та репродуктивної функції жінки, не потребує оперативного втручання; воно необхідне при наявності рудиментарного рога матки, якщо виникає скупчення менструальної крові або позаматкова вагітність. Перегородки у піхві діагностують переважно під час вагітності або пологів. Якщо вони перешкоджають народженню плода, їх розтинають.
Згідно з клінічним перебігом, аномалії розвитку матки та піхви класифікуються за двома групами:
1. Вади розвитку з однобічною затримкою відтоку менструальної крові.
2. Вади без затримки відтоку крові.
Клінічна картина цих груп є різною.
У першій групі характерна особливість — наявність менструацій, що супроводжуються болем. При ректальному обстеженні пальпується тугоеластичне утворення, яке щільно прилягає до матки або розміщене за бічною стінкою піхви. Іноді гематокольпос другої піхви діагностують як кісту гартнерового ходу піхви, а нагромадження крові у матковому розі — як пухлину матки або яєчників.
Друга група аномалій розвитку перебігає безсимптомно у дівчат, які не живуть статевим життям. У інших випадках можливі утруднення при статевих зносинах, звичне невиношування, безплідність.
Діагностувати аномалії розвитку матки досить складно, особливо з однобічною затримкою відтоку менструальної крові. Для цього використовують ультразвукове дослідження, гістеросальпінгографію та лапароскопію.
Форми подвоєння матки та піхви, якщо вони не супроводжуються порушенням відтоку менструальної крові, терапії не потребують. При однобічній затримці відтоку менструальної крові, зумовленій аномаліями розвитку матки і піхви, рекомендується хірургічне лікування з максимальним відтинанням перегородки або видаленням функціонуючого рога матки. За умов вагітності в рудиментарному розі матки виконують лапаротомію з видаленням вагітного рога.
Недорозвинення статевих органів спостерігається у тих випадках, коли їх формування призупиняється у внутрішньоутробному або дитячому віці, а в зрілому віці залишаються анатомічні та функціональні особливості статевого апарату, які відповідають дитячому організму.
Порушення диференціації статевих органів залежно від терміну внутрішньоутробного розвитку
1. Однорога матка і відсутність нирки — 2-4-й тиждень.
2. Відсутність або недорозвинення труби, рудиментарний ріг — 4-5-й тиждень.
3. Подвоєння статевого апарату — 6-8-й тиждень.
4. Різноманітні варіанти часткового подвоєння матки — 7-18-й тиждень.
5. Порушення гістогенезу матки і труб — 28-й тиждень вагітності.
Клінічна картина уроджених аномалій розвитку статевих органів у період статевого дозрівання залежить від форми вади. У всіх пацієнток при різних формах ВАРГ спостерігають жіночий фенотип з добре розвинутими вторинними статевими ознаками та нормальною функцією яєчників. Численні вади розвитку без затримки менструальної крові не проявляються в період статевого дозрівання.
Аплазію матки та піхви можна запідозрити клінічно за відсутності менструації у віці 15 років і старше або при невдалій спробі статевого життя. За даними А. Г. Курбанової, у пацієнток із синдромом Рокитанського—Кюстнера під час операції визначаються дві рудиментарні матки, розташовані пристінково, або одна матка, яка розташована в центрі малого таза. Функціонуюча рудиментарна матка при синдромі Рокитанського—Кюстнера за умов аплазії піхви виявляється у віці 17-19 років, коли з’являється біль, який щомісяця посилюється, а при ректальному обстеженні високо в порожнині малого таза пристінково на відстані 9-10 см від відхідника пальпується болісна матка округлої форми.
З-поміж численних вад розвитку геніталій у період статевого розвитку виражені клінічні прояви мають лише ті форми, які супроводжуються затримкою відтоку менструальної крові. До таких варіантів ВАРГ належать: атрезія гімена; аплазія частин або всієї піхви при функціонуючій матці (аплазія нижньої, середньої частин, двох третин або аплазія всієї піхви); подвоєння піхви та матки з частковою аплазією однієї піхви, додатковий замкнутий функціонуючий ріг матки.
Аномалії розвитку матки є результатом уродженого дефекту з порушенням процесу злиття парамезонефральних ходів. Якщо вказані анатомічні ембріональні утворення не з’єднуються, то формується uterus didelphus — дві матки, дві шийки матки та дві піхви (рис. 23).
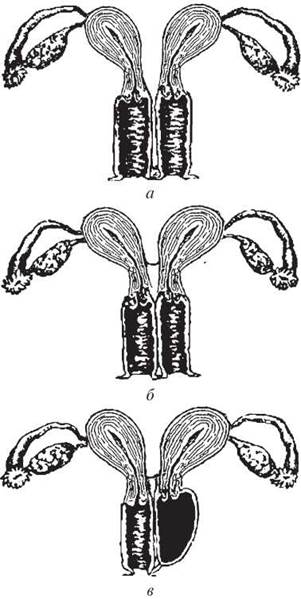
Рис. 23. Подвоєння матки та піхви: а — повне; б — неповне; в — лівобічний гематокольпос при неповному подвоєнні матки, аплазія лівої піхви
Названі варіанти ВАРГ уперше клінічно проявляються з початком менструальної функції. Для двох перших видів вад розвитку геніталій характерним є поєднання «несправжньої» аменореї та больового синдрому, для третього і четвертого (при однобічній затримці відтоку менструальної крові) — альгоменорея.
Відсутність піхви (aplasia vaginae) є вадою розвитку, яка унеможливлює здійснення менструальної, статевої та репродуктивної функцій жінки. Розвивається первинно (внутрішньоутробно) або вторинно внаслідок зарощення піхви після перенесених тяжких запальних процесів у ранньому дитинстві (віспа, дифтерія, скарлатина), дуже рідко — після тяжких пологових травм.
За умов часткової аплазії піхви виникає інтенсивніший біль, при менш сформованій частині піхви. Біль переймистий, інтенсивність його зростає, спочатку повторюється періодично, а з часом стає постійним. Розмір гематокольпосу у дівчат менший, ніж при атрезії тімена, а гематометра та гематосальпінкс утворюються частіше.
На ехограмі помітне розширення за рахунок рідкого вмісту порожнини матки. Діагностичним ехографічним критерієм часткової аплазії піхви є наявність її особливої форми, а також протяжність гематокольпосу.
Для нормально функціонуючої матки за умов повної аплазії піхви характерні скарги на біль унизу живота, який турбує з 14-15 років. На місці піхви пальпується тяж, а в малому тазі визначається збільшена, щільна, болісна матка (гематометра). Іноді збоку від матки пальпуються гематосальпінкси у вигляді утворень витягнутої форми тугоеластичної консистенції. На ехограмі збільшена матка визначається в центрі малого таза з розширеною порожниною за рахунок рідкого вмісту, з обох боків від матки — труби витягнутої форми з рідким вмістом (гематосальпінкси). Яєчники нормальних розмірів, розташовані звичайно.
Труднощі виникають при лікуванні вказаної патології — аплазії всієї піхви при функціонуючій матці, особливо якщо у матці відсутня шийка або в шийці відсутній цервікальний канал.
Лікування хірургічне — пластична операція зі створення піхви з ділянки сигмоподібної кишки. Останнім часом поширена алопластика. Спроби створити піхву різними методами рідко приводять до бажаних результатів, оскільки нерідко операція ускладнюється інфікуванням, утворюються піометра і піосальпінкси.
Подвоєння матки та піхви без затримки відтоку менструальної крові в пубертатному віці виявляється при обстеженні тільки як випадкова знахідка. Більшість аномалій матки і піхви клінічно проявляються саме в цьому віці та характеризуються повною або частковою затримкою менструальної крові, коли є друга замкнута рудиментарна піхва та другий рудиментарний ріг матки.
Формування замкнутого додаткового рудиментарного рога матки пов’язане зі значним відставанням у розвитку одного із мюллерових ходів. Якщо рудиментарний ріг має порожнину, вона може з’єднуватися з порожниною основної матки. За наявності додаткового замкнутого рога матки больовий синдром найбільш виражений. Переймистий біль з’являється через 36 міс. після менархе, він супроводжує кожну менструацію та посилюється з кожним циклом.
При додатковому замкнутому функціонуючому розі з боку від матки, розміри якої відповідають віку, пальпується щільне (різко болісне) утворення невеликих розмірів. При вагіноскопії відхилень від норми не виявляють.
Клініка перебігу захворювання є визначальною в діагностиці додаткового замкнутого функціонуючого рога матки. Ехограма показує утворення, яке за структурою нагадує матку, тісно з нею пов’язане, але розташоване асиметрично щодо дна матки. Ці ехографічні ознаки вказують на наявність другого рудиментарного рога матки.
У дівчат з подвоєнням матки та піхви, з частковою аплазією однієї з них біль супроводжує кожну менструацію, причому альгоменорея частіше проявляється через декілька місяців після менархе (після утворення гематокольпосу певних розмірів). Характер болю ідентичний тому, що й при аплазії нижньої третини піхви, але локалізується зазвичай з боку гематокольпосу.
У деяких дівчат больові відчуття під час менструації незначні, однак у них з’являються в міжменструальний період гнійні або мажучі кров’янисті виділення, що вказує на наявність отвору в перегородці між замкнутою й основною піхвами. При вагіноскопії виявляють випинання однієї зі стінок піхви за рахунок гематокольпосу частково аплазованої другої піхви, тому шийка матки не завжди доступна для огляду. Інколи визначають точкові отвори, які ведуть у другу піхву. При ректальному обстеженні вище входу в піхву пальпується пухлиноподібне утворення тістуватої консистенції з нечіткими контурами, зміщене вбік.
На ехограмі при поперечному скануванні визначають дві матки, які розташовані симетрично або асиметрично. Матки за розмірами й акустичними властивостями однакові та відповідають віковій нормі.
У деяких дівчат на боці замкнутої піхви довжина матки збільшена і порожнина розширена за рахунок рідкого вмісту — гематометри.
За допомогою ехографії встановлено, що взаємне розташування маток може бути різним: вони розміщуються пристінково або на деякій відстані одна від одної, або щільно прилягають одна до одної на всьому протязі, або з’єднані тільки в ділянці шийок; при цьому визначаються два цервікальних канали. Два яєчники розташовуються біля маток, розміри їх відповідають віковій нормі.
При ультразвуковому обстеженні ділянки піхви визначається розширення за рахунок рідкого вмісту, частіше з дрібнодисперсною зависсю, порожнина замкнутої рудиментарної піхви розташована низько, ззаду сечового міхура, ближче до стінки. Розміри гематокольпосу залежать в основному від висоти аплазії піхви та кількості нагромадженого в ній вмісту. На боці аплазованої піхви можуть візуалізуватися гематометра і гематосальпінкс.
Після оперативного відновлення прохідності замкнутої піхви при контрольному УЗД органів малого таза на ехограмі в більшості випадків визначається повне вивільнення замкнутої піхви. У деяких пацієнток виявляють залишкову порожнину, що вказує на необхідність повторного хірургічного втручання.
До форм ВАРГ, що рідко трапляються, належать вади розвитку статевих органів у поєднанні з аномаліями розвитку інших систем, найчастіше — сечовидільної. Аплазія матки та піхви інколи поєднується з тазовими нирками. У 100 % випадків — з подвоєнням матки і піхви та частковою аплазією однієї з них, на ехограмі визначається агенезія нирки на боці замкнутої піхви. Друга нирка або збільшена в розмірах і нормальної структури, або ж у ній проявляються ехографічні ознаки пієлонефриту з розширенням чашково-мискової системи, а інколи — з її подвоєнням.
У зв’язку з частим поєднанням вад розвитку статевих органів з аномаліями сечовидільної системи доцільно проводити дослідження даних систем паралельно.