Фізіологія людини - Вільям Ф. Ґанонґ 2002
Функції нервової системи
Регулювання постави і рухів
Базальні ядра
Анатомічні підстави
Термін базальні ядра звичайно стосується п’яти структур кожної півкулі мозку: хвостатого ядра, лушпини і блідої кулі, трьох великих ядерних мас, розміщених під корою великого мозку (рис. 12-9), а також зв’язаних з ними підталамічного ядра (тіло Люїса) і чорної речовини. Бліду кулю поділяють на латеральний і медіальний сегменти. Чорна речовина складається зі щільної і сітчастої частин. Ядра таламуса теж тісно пов’язані з базальними ядрами. Хвостате ядро і лушпину часто називають смугастим тілом; лушпина і бліда куля разом утворюють сочевицеподібне ядро (табл. 12-3).
Головні аферентні зв’язки, що проходять до базальних ядер, закінчуються в смугастому тілі (рис. 12-10). Вони містять волокна кірково-смугастого напряму, які відходять з усіх ділянок мозкової кори. Є також зв’язки між центросерединним ядром таламуса і смугастим тілом.
Зв’язки між окремими структурами базальних ядер охоплюють дофамінергічний чорноречовинно-смугастий шлях, тобто шлях від чорної речовини до смугастого тіла, і відповідний ГАМК-ергічний шлях від смугастого тіла до сітчастої частини чорної речовини. Хвостате ядро і лушпина пов’язані з обома сегментами блідої кулі. З латерального сегмента блідої кулі нервові волокна проходять до субталамічного ядра, від якого, відповідно, відходять волокна до обох сегментів блідої кулі і чорної речовини.
Головний шлях поширення збуджень від базальних ядер - від медіального сегмента блідої кулі через таламічний пучок до латерального вентрального, переднього вентрального і центросерединного ядер таламуса. З ядер таламуса волокна відходять до передлобової та передмоторної кори. З чорної речовини волокна теж проходять до таламуса. Ці зв’язки, а також їхні ймовірні синаптичні трансмітери показані на рис. 12-10. Є ще декілька додаткових зв’язків, спрямованих, зокрема, до повідців і верхніх горбиків. Проте найважливіші серед зв’язків базальних ядер шляхи з мозкової кори до смугастого тіла, зі смугастого тіла до медіального сегмента блідої кулі, з медіального сегмента блідої кулі до таламуса і з таламуса знову до кори, замикаючи цикл циркулювання збудження. Шлях від медіального сегмента блідої кулі до таламуса є гальмівним, тоді як шлях від таламуса до мозкової кори - збуджувальним.
Смугасте тіло містить унікальну мозаїку стріосом, утворену нервовими закінченнями в межах матриксу. Сам матрикс отримує інші закінчення. Нейрони кірково-смугастого шляху, що починаються в глибокій ділянці п’ятого шару кори, первинно закінчуються в стріосомах, тоді як нейрони, що починаються в другому і третьому, а також у поверхневій ділянці п’ятого шару, первинно закінчуються в матриксі. Нейрони, тіла яких розміщені в стріосомах, здебільшого спрямовують аксони до дофамінергічних нейронів щільної частини чорної речовини, тоді як значна кількість нейронів, тіла яких розміщені в матриксі, - до ГАМК-ергічних нейронів сітчастої частини чорної речовини. Фізіологічне значення цих зв’язків, однак, не з’ясоване.
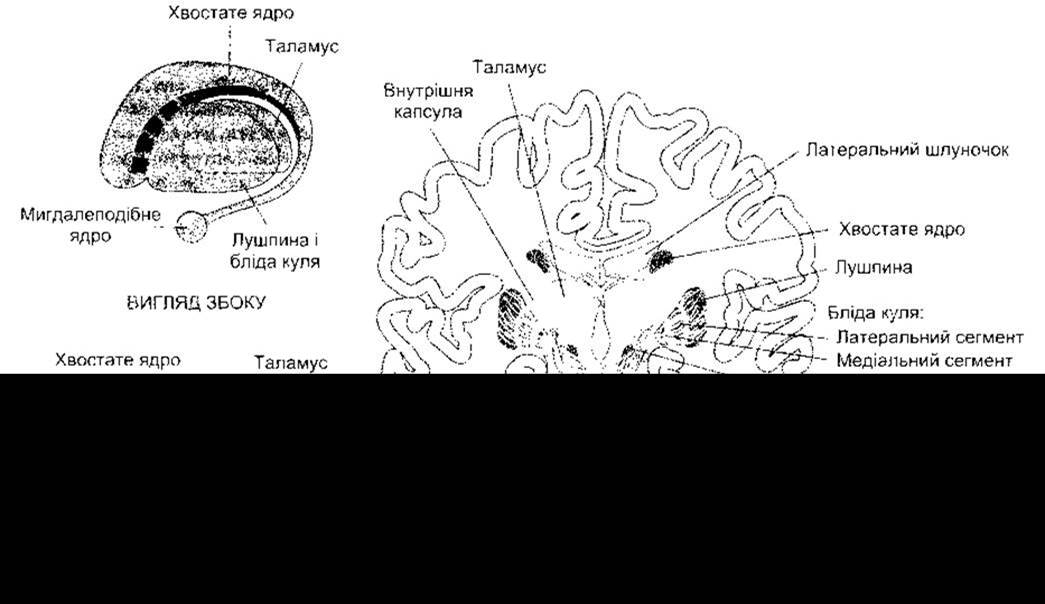
Рис. 12-9. Базальні ядра.
Особливості метаболізму
Метаболізм базальних ядер унікальний за кількістю його шляхів. У цих структурах підвищений рівень використання О2. Вміст міді в чорній речовині і суміжному голубому ядрі особливо високий. У разі хвороби Вільсона, спадкового аутосомно рецесивного розладу обміну міді, коли рівень в плазмі мідьвмісного білка церулоплазміну звичайно знижений, виникають хронічна мідна інтоксикація і значні дегенеративні зміни в сочевицеподібному ядрі.
Функція
Наші знання щодо функції базальних ядер досі обмежені. Ушкодження базальних ядер у тварин не спричинює значного ефекту. Однак з’ясовано, що нейрони базальних ядер, як і нейрони латеральних ділянок півкуль мозочка, збуджуються перед початком рухової реакції. Ці спостереження, а також детальний аналіз проявів захворювань, що виникають у базальних ядрах людини, та ефект хімічних речовин, які руйнують дофамінергічні нейрони в експериментах на тваринах (див. нижче), наводять на думку, що ці ядра беруть участь у плануванні і програмуванні рухів, або, ширше, у процесах, за допомогою яких абстрактна думка втілюється в вольову рухову реакцію (див. рис. 12-1). Імпульси з базальних ядер через таламус поширюються до ділянок, що належать до моторної кори, а кірково-спинномозковий шлях забезпечує передавання імпульсів до рухових нейронів. Крім того, збуджувальні потенціали в базальних ядрах коливаються, і треба думати, що ці коливання можуть мати подібне функційне значення, як коливання в таламо-кортикальних циклах (див. Розділ 11).
Таблиця 12-3. Базальні ядра
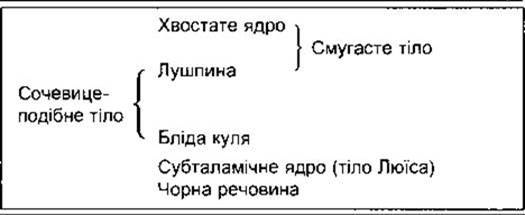
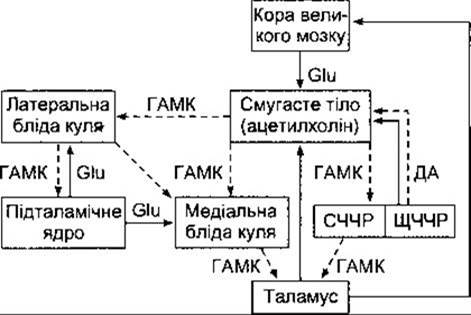
Рис. 12-10. Схематичне зображення головних зв’язків базальних ядер. Суцільними лініями позначено збуджувальні шляхи, штриховими лініями - гальмівні. Біля шляхів наведено нейротрансмітери, де вони відомі. Glu - глютамат; ДА-дофамін. Нейротрансмітер ацетилхолін утворюють інтернейрони смугастого тіла. СЧЧР - сітчаста частина чорної речовини; ЩЧЧР - щільна частина чорної речовини. Підталамічне ядро має також зв’язки зі щільною частиною чорної речовини. Для спрощення схеми цей шлях не показано.
Базальні ядра теж відіграють роль в окремих процесах пізнання, і це забезпечує хвостате ядро. Це ядро має взаємозв’язки з лобовою часткою неокортексу і, вірогідно, тому в разі його ушкодження неможливе оцінювання реакцій, що стосуються зміни і відтермінування почерговості. Крім того, ушкодження лише головки лівого хвостатого ядра і суміжної білої речовини в людини пов’язане з виникненням дисартикуляційної форми афазії, яка нагадує афазію Верніке, проте відрізняється від неї (див. Розділ 16).
Захворювання базальних ядер у людини
Цікаво, що ураження базальних ядер в експериментальних тварин спричинює невеликий ефект, тоді як патологічні процеси, що ушкоджують ці ядра в людини, призводять до значних характерних порушень рухової функції. Є два типи порушень рухової функції, пов’язані з захворюванням базальних ядер у людини: гіпокінетичний і гіперкінетичний. Гіперкінетичні порушення супроводжуються виникненням надмірних та аномальних рухів. Це, зокрема, хорея, атетоз і балізм. Гіпокінетичні порушення - акінезія і гіпокінезія.
Хорея виявляється швидкими мимовільними танцювальними рухами; атетоз - тривалими повільними судомними. Рухи в разі хореї й атетизму спочатку подібні до довільних, проте далі стають мимовільними і дезорганізованими. У випадку балізму мимовільні рухи раптові, інтенсивні, насильницькі.
Акінезія супроводжується утрудненням початку руху і послабленням спонтанних рухів. Брадикінезія полягає в сповільненні рухів.
Хвороба Гантінґтона
Першими змінами, які можна виявити в разі хвороби Гантінґтона, є ураження проміжних шипикових нейронів хвостатого ядра і лушпини сочевицеподібного ядра. Ранній симптом хвороби - уривчаста траєкторія руху руки у випадку намагання доторкнутись до певної точки, особливо помітна наприкінці руху. Згодом з’являються гіперкінетичні хорейні рухи, що поступово наростають, доки зовсім не виснажать хворого. Мова стає невиразною, а потім незрозумілою. Настає недоумство, а згодом і смерть (переважно через 10-15 років після появи симптомів). У базальних ядрах у нормі збалансовано діють три біохімічно відмінні шляхи: чорноречовинно-смугаста дофамінергічна система; внутрішньосмугаста холінергічна система і ГАМК-ергічна система, що сполучає смугасте тіло з блідою кулею і чорною речовиною. У випадку хвороби Гантінґтона ушкоджені внутрішньосмугасті ГАМК-ергічні та холінергічні нейрони. Ушкодження ГАМК-ергічних шляхів, що ведуть до латеральної блідої кулі, послаблює її інгібувальний вплив, унаслідок чого розвивається гіперкінетичність захворювання. Дегенерація чорноречовинно-смугастої дофамінергічної системи спричинює хворобу Паркінсона (див. нижче).
Хворобу Гантінґтона успадковують за аутосомно домінантним типом, і симптоми її з’являються переважно у віці від 30 до 50 років. Ненормальний ген, що зумовлює це захворювання, розміщений біля кінця короткого плеча хромосоми 4 і переважно містить 11-34 цитозин-аденін- гуанін (САО)-послідовностей, кожна, з яких кодує глютамін. У пацієнтів з хворобою Гантінґтона ця кількість кодонів зростає до 42-86 і більше. Чим більша кількість цих послідовностей, тим швидше з’являються симптоми хвороби і вона прогресує. Ген кодує гантінгтин - білок, функція якого ще не відома. Вірогідно, що втрата функції цього білка пропорційна до збільшення кількості CAG-послідовностей. Сьогодні нема засобів ефективного лікування хвороби Гантінґтона, і вона має переважно фатальні наслідки. Проте є і деяка надія. В дослідах з моделюванням хвороби на тваринах імплантація в смугасте тіло реципієнта донорської тканини смугастого тіла плоду поліпшувала пізнавальну здатність. Відомо, що в мозку хворих людей і тварин посилюється активність тканинної капсази-1 - ензиму, що регулює апоптоз. У мишей з нокаутом гена регулювального процесу апоптозу ензиму прогресування хвороби сповільнювалось.
Хвороба Гантінґтона - це одне з генетичних захворювань, кількість яких збільшується. Таким хворобам властиві збільшення кількості тринуклеотидних послідовностей. Більшість з них стосується зміни кількості CAG-послідовностей (табл. 12-4), одна - зміни кількості CGG-послідовностей, а ще одна - зміни кількості CTG-послідовностей. Усі вони пов’язані зі змінами кодувальних послідовностей у гені. Однак GAA-послідовність, що належить до некодувальних послідовностей у гені, теж пов’язана з захворюванням - атаксією Фрідрайха. Є також попередні дані про те, що збільшення кількості 12-нуклеотидних послідовностей пов’язане з виникненням рідкісної форми епілепсії.
Хвороба Паркінсона (тремтячий параліч)
У хворобі Паркінсона виявляються як гіпо-, так і гіперкінетичні ознаки. У випадках, уперше описаних Джеймсом Паркінсоном (хвороба названа його іменем), патогенетичною основою була дегенерація чорноречовинно-смугастої системи дофамінергічних нейронів. У цьому разі особливо сильно уражуються волокна, що проходять до лушпини. Паркінсонізм, що був частим пізнім ускладненням епідемій грипу в період Першої світової війни, сьогодні є спорадичною ідіопатичною формою в багатьох людей середнього і похилого віку. В цьому віці простежується стійке зменшення кількості дофаміну і дофамінових рецепторів у базальних ядрах, і прискорення цього процесу, очевидно, може бути підставою для виникнення хвороби.
Паркінсонізм може виникати теж як ускладнення після лікування транквілізаторами групи фенотіазину й іншими засобами, що блокують D2-дофамінові рецептори, або в гострій і важкій формі після ін’єкцій МФТП (рис. 12-11). Цей ефект виявлено випадково завдяки тому, що продавець лікарських засобів у Північній Каліфорнії постачав клієнтів препаратом «синтетичного героїну» кустарного виробництва, у якому був МФТП. МФТП є попередником високоактивного оксиданту МФП+, що метаболізується в астроцитах за допомогою ензиму моноамінооксидази В. У гризунів МФП швидко виводиться з головного мозку, проте в приматів його видалення набагато повільніше; він зв’язується транспортером дофаміну і надходить у дофамінергічні нейрони чорної речовини, спричинюючи їхні деструкції. Інші дофамінергічні нейрони не уражуються до такого ступеня. Тому МФТП можна використовувати для моделювання паркінсонізму в мавп, саме ця його властивість прискорила дослідження функції базальних ядер.
Гіпокінетичними проявами хвороби Паркінсона є акінезія і брадикінезія, а гігіеркінетичними - ригідність і тремор. Відсутність рухової активності й утруднення започаткування довільних рухів вражають. Простежується послаблення комбінованих рухів, таких як нормальні підсвідомі рухи, зокрема розмахування руками під час ходіння, а також збідніння міміки, що емоційно виражає думку і мову, нема численних метушливих рухів і жестів, властивих усім нам. Ригідність відрізняється від еластичності, оскільки в цьому разі знижується рівень збудження в рухових нейронах, що іннервують як м’язи-агоністи, так і м’язи-антагоністи. Пасивність рухів кінцівок поєднана з дифузною м’язовою ригідністю, що нагадує опір, який виникає під час згинання свинцевої трубки, тому її називають ригідністю свинцевої трубки. На фоні пасивності рухів виникає серія «посіпування» (ригідність зубчастого кола), проте раптової втрати резистентності, як у разі еластичності кінцівки, не відбувається. Тремор, що простежується в стані спокою і зникає під час рухів, зумовлений регулярними почерговими 8/с скороченнями м’язів-антагоністів. Згідно з загальноприйнятим поглядом в основі патогенезу хвороби Паркінсона є дисбаланс між збудженням та інгібуванням у базальних ядрах, що виникає внаслідок втрати дофамінергічного інгібування лушпини (рис. 12- 12). Зменшення інгібувального впливу на латеральну бліду кулю, що виникає в цьому разі, веде до зменшення інгібувального впливу субталамічного ядра, і це збільшує збуджувальний ефект цього ядра у медіальній блідій кулі. Унаслідок цього знижується рівень інгібувального впливу медіальної блідої кулі на таламус, а також рівень поширення збуджень до кори великого мозку.
Таблиця 12-4. Приклади хвороб, спричинених зміною кількості тринуклеотидних послідовностей
Захворювання |
Тринуклеотидна послідовність |
Дефектний білок |
Хвороба Гантінґтона |
CAG |
Гантінгтин |
Спіноцеребелярна атаксія першого типу |
CAG |
Атаксин 1 |
Спіноцеребелярна атаксія другого типу |
CAG |
Атаксин 2 |
Спіноцеребелярна атаксія третього типу |
CAG |
Атаксин 3 |
Спіноцеребелярна атаксія шостого типу |
CAG |
а1А-субодиниця Са2+-каналу |
Спіноцеребелярна атаксія сьомого типу |
CAG |
Атаксин 7 |
Зубчастоядерно- палідолюїсова атаксія |
CAG |
Атрофій |
Спінобульбарна м’язова атрофія |
CAG |
Рецептор андрогена |
Минущий Х-синдром |
CGG |
FMR-1 |
Міотонічна дистрофія |
CTG |
DM-протеїн-кіназа |
Атаксія Фрідрайха |
GAA |
Фратаксин |
Лікування
Важливим у разі обговорення патогенезу хвороби Паркінсона є питання балансу між рівнем збудження в холінергічних інгернейронах та інгібувальним дофамінергічним впливом на нейрони в смугастому тілі. Певного успіху досягнуто шляхом зменшення холінергічного впливу за допомогою антихолінергічних засобів. Сильнішого впливу можна досягти, застосувавши L-ДОФА. На відміну від дофаміну, цей його попередник проникає через гематоенцефалічний бар’єр (див. Розділ 15) і допомагає ліквідувати дефіцит дофаміну. Проте L-ДОФА перетворюють на дофамін головно дофамінергічні нейрони, а дегенерація цих нейронів триває. Отже, ефективність застосування L-ДОФА після декількох років зменшується.
Хірургічне втручання з метою руйнування медіальної блідої кулі (палідотомія) або субталамічного ядра дає змогу відновити баланс еферентних імпульсів до нормального (див. рис. 12-12). Результат, досягнутий хірургічно, можна підсилити шляхом уживлення електродів, приєднаних до розміщеного підшкірно подразника. Застосування струму високої частоти дає змогу в разі потреби тимчасово розривати цикли в ділянці кінчиків електродів.
Іншим хірургічним підходом є імплантація дофамінопродукувальної тканини в ділянку базальних ядер або біля неї. Трансплантанти тканини мозкової речовини надниркової залози або сонного гломуса того ж хворого діють як дофамінові мініпомпи, проте таким способом досягають лише короткочасного ефекту. Набагато ліпшими є наслідки трансплантації тканини смугастого тіла плоду. Є вірогідність, що клітини трансплантанта не тільки виживають у цих умовах, а й налагоджують відповідні зв’язки зі структурами базальних ядер реципієнта.
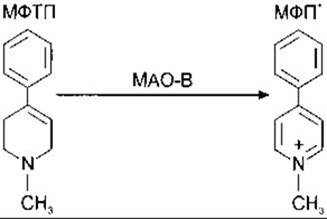
Рис. 12-11. Перетворення 1-метил-4-феніл-1,2,5,6-тетрагідропіридину (МФТП) в 1-метил-4-фенілпіридин (МФП+) моно-амінооксидазою В (МАО-В).
Водночас тривають пошуки засобів, які б запобігали явищам дегенерації дофамінергічних нейронів. Стосовно цього цікавий факт, що мутації в гені а-синуклеїну - синаптичного білка з нез’ясованою функцією - пов’язані з однією із родинних форм паркінсонізму, а тільця Леві - включення нервових клітин, характерні для всіх форм хвороби Паркінсона - саме значною мірою утворені а-синуклеїном. Врешті, тільця Леві багаті на убіквітин (див. Розділ 1), а мутація гена одного з білків, необхідних для його накопичення, пов’язана з іншою родинною формою хвороби Паркінсона. Однак зрозуміло, що потрібно ще багато досліджень, щоб зрозуміти причину дегенерації дофамінергічних нейронів, якою супроводжується хвороба Паркінсона.