Основы биохимии - А. А. Анисимов 1986
Ферменты (энзимы)
Механизм ферментативного катализа
Для протекания любой реакции необходимо, чтобы реагирующие молекулы пришли в контакт друг с другом. Однако не каждое столкновение молекул сопровождается их взаимодействием, реакция протекает только в том случае, если молекулы обладают достаточным запасом кинетической энергии. Совокупность молекул любого вещества представляет собой статистический набор молекул с различной кинетической энергией (рис. 3.5). Энергию, необходимую для достижения активированного (переходного) состояния, или тот избыток энергии по сравнению со средней энергией молекул при данной температуре, которым они должны обладать, чтобы вступить в реакцию, называют энергией активации (Еа).
В случае ферментативных реакций энергетический барьер снижается благодаря образованию фермент-субстратного комплекса, а чем меньше энергия активации, тем быстрее протекают реакции, так как вступить во взаимодействие могут молекулы с меньшим запасом энергии (рис. 3.6). Как видно из рис. 3.6, при ферментативной и неферментативной реакциях для достижения переходного состояния молекулы исходных веществ активируются, приобретают более высокий запас энергии, только после этого они могут претерпевать превращение в продукты реакции, но Еа в случае ферментативной реакции ниже.
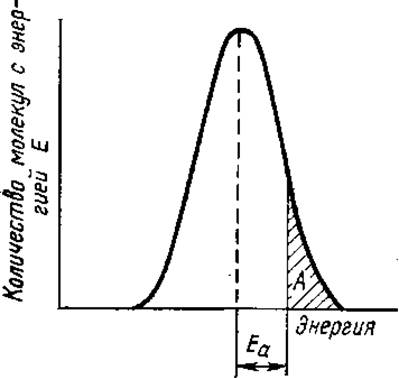
Рис. 3.5. Распределение кинетической энергии в популяции молекул:
А — молекулы, вступающие в реакцию (активированные молекулы), Еа — минимальная энергия активации
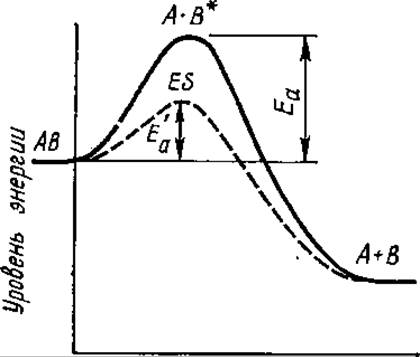
Рис. 3.6. Профили энергии реакции: Еа и Е'а — энергия активации неферментативной и ферментативной реакций, AB — исходное вещество, А+В — продукты реакции, А⋅В* — активированные молекулы, ES — активированный комплекс
Таким образом, большие скорости ферментативных реакций являются в конечном счете результатом снижения энергии активации катализируемых реакций. Именно благодаря тому, что биологические катализаторы снижают энергию активации, ферментативные реакции протекают с высокой скоростью при относительно низкой температуре.
Снижение энергии активации при ферментативном катализе имеет некоторую связь с многостадийностью этих реакций. Они протекают не в один этап, а ступенчато, через несколько промежуточных реакций. Активационный барьер реакции при этом разбивается на несколько более низких барьеров каждой промежуточной реакции, преодолеть которые реагирующим молекулам легче, чем один большой барьер.
Ферментативный катализ имеет признаки как гомогенного, так и гетерогенного катализа, протекающего на границе раздела двух фаз. В каталитическом действии ферментов можно выделить 3 стадии: 1) присоединение молекулы субстрата (S) к ферменту (Е), 2) превращение субстрата, 3) отделение конечных продуктов реакции (Р) от фермента. Простейшая схема ферментативной реакции записывается следующим образом:
![]()
Наиболее быстрой обычно является первая стадия реакции, медленной — вторая. На 1-й стадии реакции происходит образование фермент-субстратного комплекса (ES, ФСК), в результате чего структура и свойства молекулы субстрата меняются, образуются его переходные формы. Это является главной предпосылкой ускорения процесса его превращения в катализируемой реакции.
Образование ФСК становится возможный благодаря определенному сродству ферментов к своим субстратам. Наиболее четко мысль о таком соответствии была высказана Э. Фишером (1890) при объяснении специфичности действия ферментов. Он сравнивал фермент и субстрат с ключом и замком и полагал, что фермент подходит к своему субстрату как ключ к замку. В настоящее время не вызывает сомнения, что между пространственными структурами субстрата и активного центра фермента существует стерическое соответствие, однако это соответствие, как будет показано дальше, не абсолютное. В образовании фермент-субстратного комплекса принимают участие ионные, водородные связи и гидрофобные взаимодействия. Возникновение временных ковалентных связей между ферментом и субстратом возможно в ряде ферментативных реакций только на 2-й стадии — при превращении субстрата и образовании промежуточных и переходных состояний. ФСК очень лабильны: существуют лишь доли секунды, тем не менее к настоящему времени удалось получить ряд ФСК.
Образование ФСК создает предпосылки высокой каталитической активности. Установлено, что при образовании ФСК молекулы фермента и субстрата не только сближаются, но и определенным образом ориентируются друг относительно друга. Между структурой субстрата и активного центра фермента кроме стерического соответствия существует и топохимическое, при котором обеспечивается взаимодействие узнающих групп фермента (связывающая зона) и узнаваемых групп субстрата. Связывание фермента и субстрата обычно многоточечное, причем чем выше специфичность фермента, тем больше точек узнавания.
Немаловажным фактором, вносящим определенный вклад в возрастание скорости ферментативных реакций, является увеличение на несколько порядков времени контакта реагирующих молекул в результате многоточечного связывания субстрата (ов) ферментом. Время, в течение которого длится контакт молекул при столкновении в случае чисто химической реакции, равно приблизительно периоду тепловых колебаний молекул (10-13—10-12 с). За это время не всегда успевает произойти химическая реакция.
Определенный вклад в увеличение скорости ферментативных реакций вносит индуцированное соответствие фермента субстрату. Согласно теории индуцированного соответствия, высказанной впервые Д. Кошландом, у многих ферментов в отсутствие субстратов функциональные группы активных центров ориентированы таким образом, что оптимального взаимодействия их с комплементарными группами субстрата не происходит. Вхождение субстрата в активный центр фермента вызывает конформационное изменение в ферменте, при котором функциональные группы фермента занимают положение, оптимальное для протекания каталитического процесса. Доказательством конформационных изменений фермента при связывании субстрата является отличие рентгенограмм свободного фермента и в присутствии специфического ингибитора. Наличие конформационных изменений при связывании субстратов и их аналогов хорошо показано для карбоксипептидазы А, лизоцима, гидролизующего полисахариды клеточных стенок бактерий. Например, у карбоксипептидазы А в присутствии аналога субстрата наблюдается смещение ряда аминокислот в активном центре (тир-248 смещается приблизительно на 1,2 нм, арг-145 и глу-270 — на 0,2 нм).
При оптимальном связывании субстрата (ов) с ферментом образуется продуктивный фермент-субстратный комплекс, т. е. такой комплекс, который через ряд промежуточных стадий дает продукт(ы) реакции. Эти процессы протекают на 2-й стадии ферментативной реакции. Быстрому протеканию ферментативной реакции способствует то, что при взаимодействии субстрата с ферментом индуцируется напряжение разрываемых связей в субстрате (деформация или дестабилизация их), т. е. разрываемые связи становятся менее стабильными, чем в свободном субстрате.
Увеличение скорости реакции под влиянием ферментов происходит и благодаря большей гидрофобности среды активного центра по сравнению с окружающим раствором, в такой среде наблюдается десольватация заряженного субстрата, приводящая к дестабилизации разрываемой связи.
Важной особенностью ферментативных реакций является и то, что превращение субстрата протекает как полифункциональный катализ. Полифункциональность обеспечивается разнообразием аминокислотных остатков белковой части фермента и групп кофакторов в активном центре; на превращающуюся химическую связь субстрата одновременно или в результате серии последовательных атак воздействует несколько групп фермента. В результате этого происходит поляризация превращающейся связи и затем ее разрыв.
Многие группы в активных центрах ферментов функционируют как обобщенные кислоты или основания, воздействуют на субстрат, активируют его и тем самым увеличивают скорость катализа. Обобщенные кислоты, согласно Бренстеду, — это любые доноры протонов, а основания — акцепторы протонов. Особенно эффективен обобщенный кислотно-основной катализ. Он дает увеличение скорости в 10—100 раз. В качестве обобщенных кислотно-основных катализаторов функционируют в активном центре боковые радикалы таких аминокислот, как, например, глу, асп, гис, лиз, тир и др. В протонированной форме они являются кислотными катализаторами, в непротонированной — основными.
Для ферментативных реакций большое значение имеет и электрофильно-нуклеофильный катализ. В активных центрах некоторых ферментов есть электрофильные и нуклеофильные группы, принимающие участие в акте катализа. Электрофильная группа — это акцептор электронной пары (кислота Льюиса), нуклеофильная — донор электронной пары (основание Льюиса).
Нуклеофильные группы ферментов вступают в реакции нуклеофильного замещения, что приводит к образованию ковалентных промежуточных соединений, — ковалентный катализ. Нуклеофильная группа становится на место замещаемой группы, образуется ковалентный интермедиат; он неустойчив и легко распадается на продукты реакции. Сильным нуклеофилом является имидазольная группа гис, поэтому химическая модификация гис в составе активного центра приводит к инактивации ферментов. К нуклеофильным группам относятся также ОН-группа сер, SH-группа цис. Примерами электрофильных групп являются ионы металлов — Zn2+-, Fe3+ и др.
Известно значительное количество ферментов, образующих с субстратами или промежуточными продуктами их превращения ковалентные интермедиаты, при этом определенные группы активного центра подвергаются ковалентной модификации за счет субстратов. Например, модификация сер в химотрипсине, субтилизине и плазмине происходит с образованием эфира карбоновой кислоты 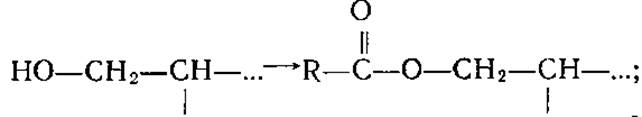 в щелочной фосфатазе образуется эфир фосфорной кислоты
в щелочной фосфатазе образуется эфир фосфорной кислоты ![]()
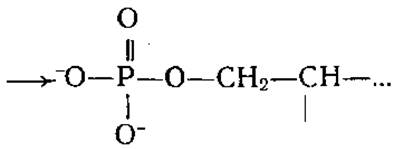 В папаине и глицеральдегидфосфатдегидрогеназе цис модифицируется в тиоэфир карбоновой кислоты
В папаине и глицеральдегидфосфатдегидрогеназе цис модифицируется в тиоэфир карбоновой кислоты ![]()
в трансальдолазе и пиридоксальзависимых ферментах лиз дает ковалентный интермедиат с субстратами — шиффово основание H2N—(СН2)4—![]()
Образование ковалентных интермедиатов избирательно увеличивает вероятность протекания определенной реакции, поскольку ковалентно связанный интермедиат обладает ограниченной подвижностью и может поэтому занимать более благоприятное положение для завершения реакции по отношению к соответствующим группам фермента. Ускорение ферментативной реакции в результате образования ковалентного интермедиата возрастает в 102—103 и более раз.
Специфический кислотно-основной катализ, т. е. активация субстратов под влиянием повышенных концентраций ионов Н3О+(Н+) и ОН-, в биохимических реакциях встречается редко, этот тип реакций широко распространен в органической химии. Таким образом, большие скорости ферментативных реакций возможны благодаря наличию целого ряда эффектов: эффект сближения и ориентационный эффект, имеющие место благодаря стеричсскому и топохимическому соответствию фермента и субстрата, индуцированное соответствие структуры активного центра переходному состоянию субстрата и возникновение «напряжения» в субстрате, полифункциональность и многостадийность катализа, особые физико-химические условия среды активного центра и как результат всего — снижение энергии активации катализируемой реакции. Все эти эффекты обусловлены сложным строением молекул ферментов. Уникальное строение активного центра фермента обеспечивает и его высокую специфичность.
Структура активного центра, механизм связывания субстрата, каталитические свойства и первичная структура хорошо изучены у таких гидролитических ферментов, как химотрипсин, РНКаза, лизоцим, карбоксипептидаза А. Описан также механизм действия каждого из этих ферментов. Ниже в качестве примеров кратко рассмотрены представления о механизме некоторых ферментативных реакций. В активном центре химотрипсина важная роль в акте катализа принадлежит сер-195 и гис-57. Расстояние между ОН-группой сер и третьим атомом имидазола гис равно 0,3 нм, между ними возникает водородная связь, что увеличивает реакционную способность гидроксильной группы сер. Первый атом азота гис-57 образует водородную связь с СООН-группой асп-102, также входящей в активный центр химотрипсина, в результате чего образуется система с переносом заряда, состоящая из асп-102, гис-57 и сер-195 (рис. 3.7, стадия I).
Анион ОН-группы сер осуществляет нуклеофильную атаку углерода расщепляемой пептидной связи субстрата, образуется ацил-фермент, и вслед за этим освобождается первый продукт реакции — аминный продукт (стадия II). Далее происходит гидролиз ацилфермента, образование 2-го продукта реакции, а фермент возвращается в исходное состояние (стадии III, IV).
При связывании лизоцима с субстратом — полисахаридом клеточных стенок бактерий, состоящим из большого числа дисахаридных единиц (N-ацетилглюкозамин— N-ацетилмурамовая кислота), происходит вынужденный контакт. Он сводится к небольшому перемещению (~0,07 нм) некоторых боковых аминокислотных групп внутри активного центра. При связывании субстрата одно из колец гексоз, располагающееся против каталитических групп активного центра, переходит в напряженное состояние (из конформации кресла в конформацию полукресла), что способствует ускорению гидролиза гликозидной связи, в которой участвует гексоза в напряженной конформации (рис. 3.8).
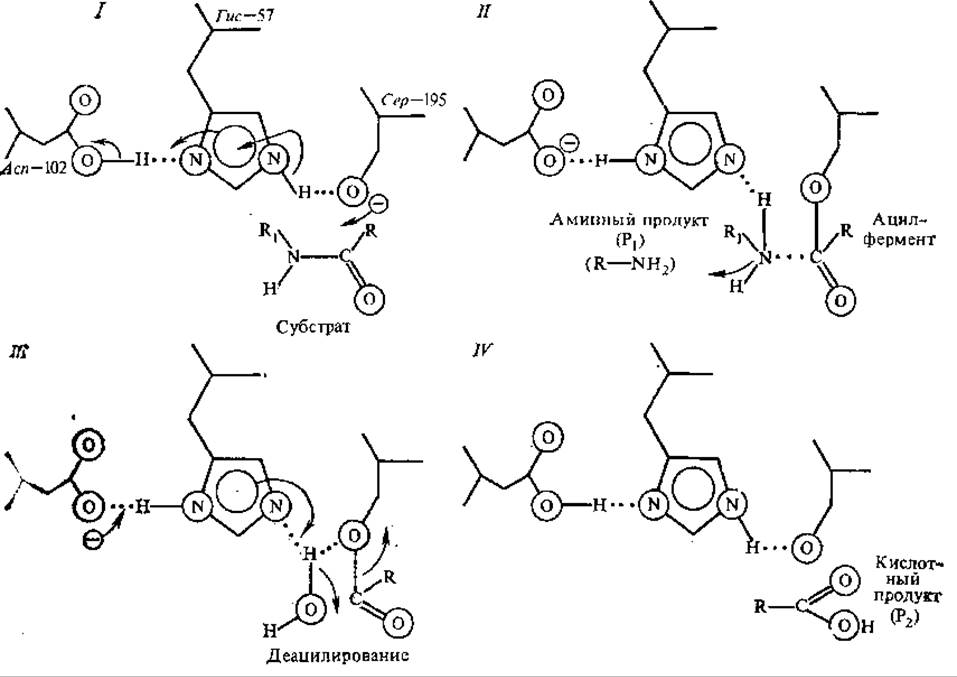
Рис. 3.7. Последовательность стадий катализа химотрипсином (пояснение см. в тексте)
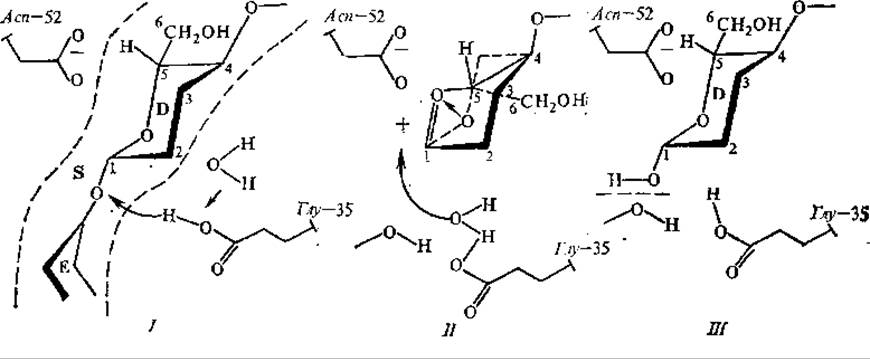
Рис. 3.8. Предполагаемый механизм действия лизоцима: I, IІ, III — стадии реакции, D, Е — остатки гексоз субстрата (S)
Каталитическими остатками активного центра являются глу-35 и асп-52, причем глу-35 находится в гидрофобном окружении в протонированном состоянии, а асп-52 — в гидрофильном окружении и ионизирован (рис. 3.8, стадия I). Глу-35 выступает донором протона для кислорода разрываемой гликозидной связи. Вместе с разрывом связи образуется карбониевый ион (С+) гексозы, стабилизируемый отрицательным зарядом на асп-52 (стадия II). Далее происходит гидролиз: ОН-группа воды присоединяется к карбониевому остатку, протон воды — к глу-35, и реакция завершается (стадия III), В увеличение скорости реакции, катализируемой лизоцимом, дают вклад следующие эффекты: напряженная конформация субстрата, вынужденный контакт, обобщенный кислотно-основной катализ при участии глу-35, стабилизация карбониевого иона асп-52.
Одна из функций, которую выполняют металлы в ферментах, заключается в том, что они выступают в роли электрофильных агентов. Так, в активном центре карбоксипептидазы А, содержащем ион Zn2+, последний представляет собой электрофильный агент, оттягивающий электроны от пептидной связи субстрата, облегчая ее гидролиз (рис. 3.9).
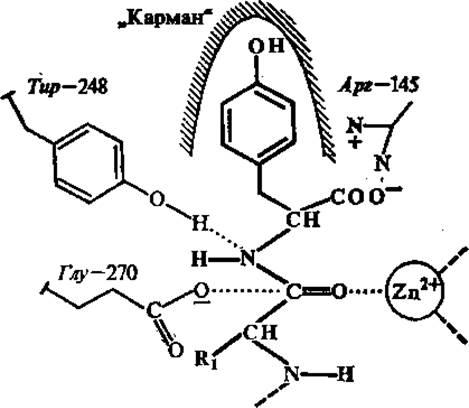
Рис. 3.9. Связывание С-концевого фрагмента субстрата (показан более толстыми линиями) в активном центре карбоксипептидазы А:
Zn2+ — электрофильный агент, воздействующий на расщепляемую пептидную связь